
Научная электронная библиотека
Монографии, изданные в издательстве Российской Академии Естествознания

Мутации гена FMR1
Ген FMR1, с которым связан синдром FRAXA, имеет особую форму мутации. Данная мутация представляет собой увеличение (экспансию) числа тринуклеотидных повторов цитозин-гуанин-гуанин (CGG) в регуляторной области гена FMR1 (более 200 повторов), сопровождающееся аномальным метилированием [Юров и др., 2005 а; Verkerk et al., 1991]. Следствием последнего является нарушение экспрессии гена FMR1. Такое состояние гена FMR1 носит название полной мутации [Kremer et al., 1991]. Нестабильные аллели, которые предшествуют возникновению аллелей с полной мутацией, носят название премутационных и связаны с фенотипами, отличными от синдрома FRAXA. К этим фенотипам относят преждевременное нарушение функции яичников (Premature ovarian failure, POF) и синдром тремора и атаксии (Fragile X associated tremor/ataxia syndrome, FXTAS, OMIM 300623).
Молекулярно-генетические исследования показали, что ген FMR1 (fragile X mental retardation gene 1) расположен в участке Xq27.3 длинного плеча хромосомы Х, имеет величину около 38 тыс. пн, включает в себя 17 экзонов и кодирует мРНК длиной 3,9 тыс. пн [Bardoni et al., 2001]. Продуктом экспрессии данного гена является белок FMRP (fragile X mental retardation protein), который преимущественно локализуется в цитоплазме клеток, где связан с полирибосомами, присоединенными к эндоплазматическому ретикулуму, и свободными рибосомами. FMRP найден практически во всех тканях, но наивысшие уровни его концентрации наблюдаются в мозге (особенно в клетках Пуркинье мозжечка, нейронах гиппокампа и базальных отделах лобных долей), тестикулах, пищеводе, легких и почках [Khandjian, 1999]. FMRP необходим для связывания и транспортировки из ядра в цитоплазму молекул мРНК, требующихся для развития дендритов и функционирования синапсов. Кроме того, он является регулятором трансляции специфических мРНК в синапсах нейронов [Oostra, Willemsen, 2009].
Синдром FRAXA стал первым известным заболеванием, возникающим вследствие экспансии числа тринуклеотидных повторов. Позднее было открыто более 20 неврологических заболеваний с подобным
механизмом: хорея Гентигтона, спинальная и бульбарная мышечная атрофия Кеннеди, 6 форм спиноцеребеллярной атаксии и др. [Brouwer et al., 2009]. Фенотип зависит от числа CGG повторов в промоторе гена FMR1. В зависимости от числа CGG-повторов ген FMR1 имеет следующие варианты (рис. 5). В норме число повторов CGG в гене FMR1 составляет от 5 до 44 и каждые 9–10 повторов прерываются триплетами AGG, что обеспечивает стабильность числа CGG-повторов. Наиболее часто в популяции (98 %) встречаются аллели с числом CGG-повторов 28 и 30.
Промежуточные аллели – 45–54 CGG-повторов. Клиническое значение промежуточных аллелей остается неясным. Риск нестабильности числа повторов при передаче потомству возрастает, если их количество более пятидесяти и непрерываемое число повторов превышает 35.
Премутационные аллели – число повторов варьирует в пределах от 55 до 200. Из-за нестабильности таких аллелей женщины-носительницы премутации имеют высокий риск увеличения числа CGG-повторов до более 200 и рождения ребенка с синдромом FRAXA [Brouwer et al., 2009]. Носительство премутации встречается у одного из 250–810 мужчин и одной из 130–260 женщин [Hagerman, 2008]. Исследования премутаций у индивидуумов разных рас и национальностей обнаружили, что они наиболее распространены на среднем Востоке и наиболее редки – в Китае [Toledano-Alhadef et al., 2001]. Переход премутации в мутацию может происходить только в материнском гаметогенезе при делении и созревании половых клеток [Sutherland et al., 1991]. Наименьшее известное число CGG-повторов, которое привело к их экспансии и появлению у потомства полной мутации, равно 56 [Fernandez-Carvajal et al., 2009]. Премутационные аллели неметилированы и транскрипционно активны. У носителей премутации был обнаружен повышенный уровень мРНК гена FMR1 в лейкоцитах, а также сниженный уровень белка FMRP, который был тем ниже, чем выше число CGG-повторов. По аналогии с другими заболеваниями экспансии тринуклеотидных повторов была предложена модель патогенеза заболеваний, ассоциированных с премутацией гена FMR1, который может быть связан с токсическим влиянием мРНК на клетки головного мозга [Oostra, Willemsen, 2009]. Это предположение подтверждается присутствием внутриядерных включений в нейронах и астроцитах, а также клетках периферических нервных ганглиев и тестикул у больных с синдромом тремора и атаксии [Hagerman, Hagerman, 2004; Gokden et al., 2008].
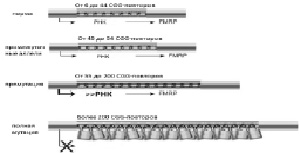
Рис. 5. Влияние экспансии CGG-повторов в гене FMR1 на его транскрипцию. Представлены 4 класса аллелей гена FMR1. Уровень транскрипции обозначен толщиной черной линии со стрелкой. Повышенная транскрипция характерна для премутации гена FMR1. Замками обозначено метилирование CGG-повторов в случае полной мутации [модифицировано из Oostra, Willemsen, 2009]
Полная мутация гена FMR1 (количество повторов превышает 200, а в некоторых случаях – 1000) с высокой вероятностью приводит к тому, что промотор гена подвергнется метилированию. Это препятствует экспрессии гена FMR1 (мРНК и FMRP не образуются), приводя к фенотипическим проявлениям синдрома FRAXA. Таким образом, аллели гена FMR1 при синдроме FRAXA являются гиперметилированными и транскрипционно неактивными. Представляют интерес единичные наблюдения за интеллектуально сохранными мужчинами с неметилированной полной мутацией, которые подтверждают то, что увеличение CGG-повторов в ряде случаев может не препятствовать экспрессии гена [Юров и др., 2005 а]. Было показано, что частота аллелей с полной мутацией в популяции составляет 1:2500, в то время как их частота в сочетании с заболеванием 1:3600, поскольку некоторые индивидуумы с полной мутацией, особенно женщины, не имеют значительного снижения интеллекта [Hagerman, 2008]. У индивидуумов с экспансией CGG-повторов в 15 % случаев может наблюдаться мозаицизм длины повторов и их метилирования. Помимо экспансии тринуклеотидных повторов в 1 % случаев синдрома FRAXA выявляют делеции, а также точковые мутации в гене FMR1. Фенотип синдрома FRAXA связан с нарушением структуры хромосомы Х: при кариотипировании клеток, выросших в культуральных питательных средах с пониженным содержанием фолиевой кислоты, обнаруживается ломкий участок хромосомы Х в участке Хq27.3 [Вехова и др., 1992; Ворсанова и др., 1998а].