
Научная электронная библиотека
Монографии, изданные в издательстве Российской Академии Естествознания

6.6. Структурный анализ данных термогравиметрического анализа
Выше было показано (см. раздел 6.2), что пленки ПК, полученные из растворов в разных растворителях, имеют разную структуру, и это различие сохраняется для их расплавов (эффект памяти). Различие структуры расплава ПК, характеризуемой фрактальной размерностью макромолекулярного клубка Df, оказывает сильное влияние на процессы термоокислительной деструкции этих расплавов (при Тст = 513 К) (см. главу 7). Исследования методом термогравиметрического анализа (ТГА) подтвердили эти результаты [77].
Экзотермический пик ДТА при ТДТА = 633 ¸ 653 К приписан образованию и реакции летучих продуктов (распад гидроперекисей, гидролиз, алкоголиз). Это соответствует химическому механизму автоускоренного режима ПК, связанному с реакцией подвижных радикалов (см. предыдущий раздел). Величины ТДТА для пленок ПК, полученных из растворов в тетрагидрофуране и 1,4-диоксане (автоускоренный режим) равны 683 и 638 К, что хорошо согласуется с цитированными выше данными [77].
Для автозамедленного режима окисления авторы [77] предположили механизм термоокислительной деструкции цепи. Для пленок ПК, полученных из растворов в хлористом метилене, хлороформе и хлорбензоле, величины ТДТА равны 740 ¸ 754 К, что вновь согласуется с литературными данными [37], где для этого режима ПК получено ТДТА > 723 K.
Расчет энергии активации ![]() термоокислительной деструкции по данным ТГА (метод Коутса-Редферна [78]) и ДТА (метод Мейкока [78]) показал близкое соответствие величин
термоокислительной деструкции по данным ТГА (метод Коутса-Редферна [78]) и ДТА (метод Мейкока [78]) показал близкое соответствие величин ![]() и
и ![]() , полученных этими методами (среднее расхождение
, полученных этими методами (среднее расхождение ![]() и
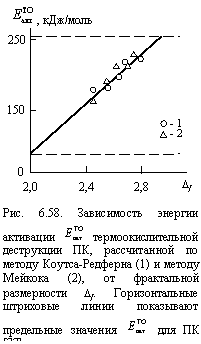
и ![]() составляет 6 %, табл. 6.3). на рис. 6.58 показана зависимость указанных энергий активации от Df, а горизонтальные штриховые линии указывают предельные значения
составляет 6 %, табл. 6.3). на рис. 6.58 показана зависимость указанных энергий активации от Df, а горизонтальные штриховые линии указывают предельные значения ![]() для ПК, полученные разными авторами и разными методами, цитированные в [37]. Как и для ПААСО (рис. 6.35), получено линейное увеличение
для ПК, полученные разными авторами и разными методами, цитированные в [37]. Как и для ПААСО (рис. 6.35), получено линейное увеличение ![]() по мере роста Df, причем указанные выше предельные литературные значения
по мере роста Df, причем указанные выше предельные литературные значения ![]() хорошо согласуются с предельными значениями Df (2 £ Df < 3 для d = 3).
хорошо согласуются с предельными значениями Df (2 £ Df < 3 для d = 3).
Рассмотрим изменение порядка реакции окисления n по мере вариации структуры макромолекулярного клубка, характеризуемой Df. В рамках фрактальной модели кинетики реакций величину n можно определить согласно уравнениями (1.70), (1.72) и (5.18). Расчет согласно этим уравнениям показал, что по мере роста Df от 2,45 до 2,74 величина n снижается от 3,63 до 3,02 (табл. 6.3). Величину n также можно определить из аппроксимационного соотношения Дойля [78]:
![]() , (6.77)
, (6.77)
где am - степень преобразования на кривой ТГА, соответствующая пику ДТА.
Таблица 6.3
Экспериментальные и расчетные параметры кривых ТГА и ДТА для пленок ПК, полученных из растворов в разных растворителях [77]
|
Растворитель |
Df |
ТДТА, К |
|
|
n |
|
|
|
Хлористый метилен |
2,45 |
754 |
183 |
164 |
3,63 |
0,60 |
0,61 |
|
Хлороформ |
2,54 |
745 |
212 |
195 |
3,40 |
0,66 |
0,62 |
|
Хлорбензол |
2,60 |
740 |
200 |
220 |
3,27 |
0,67 |
0,59 |
|
Тетрагидрофуран |
2,69 |
683 |
213 |
216 |
3,10 |
0,53 |
0,58 |
|
1,4-диоксан |
2,74 |
638 |
230 |
233 |
3,02 |
0,53 |
0,58 |
В табл. 6.3 приведено сравнение величин am, полученных экспериментально по кривым ТГА и ДТА ![]() и рассчитанных по уравнению (6.77) при условии оценки n согласно уравнению (5.18)
и рассчитанных по уравнению (6.77) при условии оценки n согласно уравнению (5.18) ![]() . Как можно видеть, между этими значениями am получено хорошее соответствие (среднее расхождение составляет 7 %), что подтверждает корректность сделанной выше оценки n в рамках фрактальной кинетики реакций [79].
. Как можно видеть, между этими значениями am получено хорошее соответствие (среднее расхождение составляет 7 %), что подтверждает корректность сделанной выше оценки n в рамках фрактальной кинетики реакций [79].
![]() , (6.78)
, (6.78)
где a - степень превращения.
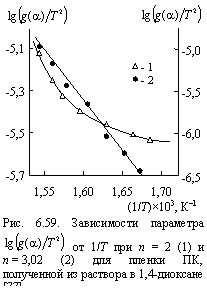
На рис. 6.59 приведено сравнение зависимостей ![]() от 1/Т при n = 2 (евклидово пространство) и n = 3,02 (фрактальное пространство) для пленки ПК, полученной из раствора в 1,4-диоксане. Как можно видеть, указанная зависимость линейна только для n = 3,02, что подтверждает корректность выбора этой величины n.
от 1/Т при n = 2 (евклидово пространство) и n = 3,02 (фрактальное пространство) для пленки ПК, полученной из раствора в 1,4-диоксане. Как можно видеть, указанная зависимость линейна только для n = 3,02, что подтверждает корректность выбора этой величины n.
В настоящее время известно большое число кинетических уравнений, описывающих процессы термоокислительной деструкции полимерных материалов, что затрудняет выбор оптимальной модели для описания этого процесса в случае конкретной полимерной матрицы. Поэтому представляет интерес исследование возможности определения оптимальных параметров процесса термоокислительной деструкции ПАр и композитов на его основе, наполненных короткими органическими волокнами (угленом и вниивлоном) обработкой экспериментальных данных ТГА с привлечением математических моделей различных гетерогенных реакций [80].
Кинетический анализ данных ТГА композитов на основе ПАр основан на использовании уравнения [80]:
![]() , (6.79)
, (6.79)
где a - степень превращения; t - время; kd - константа скорости термоокислительной деструкции; f(a) - алгебраическая функция, описывающая механизм процесса.

На рис. 6.60 приведена зависимость ![]() (Df) для ПАр и указанных органопластиков, из которой вновь следует рост
(Df) для ПАр и указанных органопластиков, из которой вновь следует рост ![]() по мере повышения Df, т.е., повышения плотности макромолекулярного клубка в расплаве. На рис. 6.60 горизонтальными штриховыми линиями указаны предельные значения
по мере повышения Df, т.е., повышения плотности макромолекулярного клубка в расплаве. На рис. 6.60 горизонтальными штриховыми линиями указаны предельные значения ![]() для ПАр согласно литературным данным [37]. Как видно, они хорошо согласуются с предельными значениями функции
для ПАр согласно литературным данным [37]. Как видно, они хорошо согласуются с предельными значениями функции ![]() (Df) при Df = 2 и 3. Кроме того, интервал температур пиков ДТА (673 ¸ 723К) указывает на автозамедленный режим окисления [81].
(Df) при Df = 2 и 3. Кроме того, интервал температур пиков ДТА (673 ¸ 723К) указывает на автозамедленный режим окисления [81].
Расчет порядка реакции n для ПАр и органопластиков на его основе согласно уравнениям (1.70), (1.72) и (5.18) показал снижение n от 4,97 до 3,27 по мере роста Df от 2,13 до 2,60, что полностью согласуется с данными для ПК [77]. Сравнение величин am, полученных экспериментально по кривым ТГА и ДТА (0,58 ¸ 0,70) и рассчитанных по уравнению (6.77) (уравнению Дойля) при условии оценки n согласно уравнению (5.18) (0,614 ¸ 0,668) показало их хорошее соответствие (среднее расхождение составляет 5 %). Таким образом, данные ТГА и ДТА подтверждают сильное влияние структуры расплава полимерной матрицы органопластиков на процессы их окисления [81].
Как было указано в разделе 6.2 и будет подробно рассмотрено в следующей главе, авторы [71] предложили структурную модель перехода от одного типа кинетических кривых окисления к другому в рамках фрактального анализа. Для двух гетероцепных полимеров - ПК и ПААСО - было показано, что в случае фрактальной размерности макромолекулярного клубка в расплаве Df меньше 2,77 реализуется автозамедленный режим окисления, а в случае Df ³ 2,77 - автоускоренный. Кроме того, изменение типа кинетических кривых связано с вариацией химических механизмов окисления, выраженной, в частности, изменением типа случайного блуждания [55], которым моделируются подвижные реагенты. В работе [82] был выполнен теоретический расчет температуры 5%-й потери массы в испытаниях ТГА для указанных выше полимеров в рамках фрактального анализа и выяснена взаимосвязь типа кинетической кривой с механизмом диффузии подвижных реагентов (медленная и быстрая диффузия).
Как показано в главе 2, в основу указанного выше деления механизмов диффузии положена зависимость смещения подвижного реагента от времени, выраженная соотношением (2.56). В этом соотношении показатель b равен ½ для классического случая, для медленной диффузии b < ½ и для быстрой - b > ½ [83]. В рамках теории дробных производных была показана взаимосвязь Df и b, которая аналитически выражается следующим образом [83]:
![]() (6.80)
(6.80)
для медленной диффузии и
![]() (6.81)
(6.81)
для быстрой.
Далее по аналогии с уравнением, описывающим процессы диффузии газов в полимерных мембранах (см. уравнение (2.60)), было использовано следующее уравнение для оценки температуры 5%-й потери массы Т5% в испытаниях ТГА [82]:
Df = c(T5% - Tc)b, (6.82)
где с - константа, Тс - температура стеклования полимера.

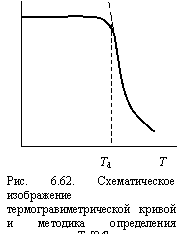
Как известно [84], температура интенсивной термодеструкции Td характеризует термостойкость полимеров. В качестве характеристики термостойкости согласно [85] принимается «предельная температура, при которой происходит химическое изменение полимера, отражающееся на его свойствах». Термостойкость определяется с помощью ТГА. В дальнейшем под Тd будет пониматься температура, определенная по пересечению касательных к двум ветвям термогравиметрической кривой (см. рис. 6.62) [84].
![]()
Важность параметра Td предопределила достаточно обширную литературу, посвященную исследованию зависимости величины Td от характеристик полимеров [84-88]. Тем не менее, эти исследования учитывают взаимосвязь Td только с химическим строением полимера в том или ином варианте: наличие «слабых звеньев» [86], тех или иных групп в полимерной цепи [87], дефектов полимерной цепи [88] и т.д. Однако физическая структура полимерного материала во всех этих исследованиях не учитывалась, хотя авторы [77] показали, что для пленок ПК, полученных из разных растворителей (что предполагает неизменность химического строения полимера) различие Td может достигать 120 К. Этот результат предполагает сильное влияние структуры полимера при температуре испытаний на его термостойкость. В этом смысле наиболее удобными объектами для исследования являются полимерные пленки одного и того же полимера, полученные из разных растворителей, и полимерные композиты. Для этих классов полимерных материалов возможны достаточно большие вариации их физической структуры без изменения химического строения полимера (полимерной матрицы) [77; 89]. Поэтому авторы [90] выполнили исследование влияния структуры полимерных материалов на их термостойкость и получили аналитическую взаимосвязь Td и структурных характеристик на примере пленок ПК, полученных из растворов в пяти разных растворителях (см. табл. 7.1), и углепластиков на основе фенилона [91]. Указанные углепластики получены технологией предварительного смешения компонентов во вращающемся электромагнитном поле, что позволяет вариацией продолжительности смешения изменять структуру полимерной матрицы в достаточно широких пределах (Df = 2,29 ¸ 2,57).
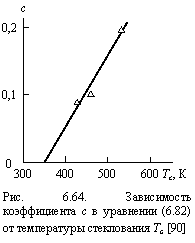
Уравнение (6.82) определяет три фактора, влияющие на термостойкость полимерных материалов: химическое строение полимера, характеризуемой величиной Тс, структуру полимерного расплава Df и тип (интенсивность) диффузии оксиданта, связанный со структурой и характеризуемый показателем b (уравнения (6.80) и (6.81) [90].
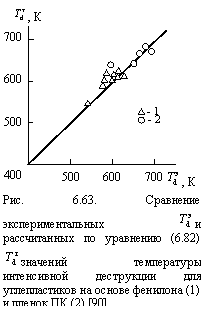
Как показали оценки величины с согласно методу наилучшего совмещения теории и эксперимента, эта константа имеет разные значения для разных полимеров и разных типов диффузии. Так, в случае углепластиков на основе фенилона с = 0,20 для быстрой и 0,50 для медленной диффузии и в случае ПК с = 0,093 и 0,30, соответственно. Cравнение экспериментальных и рассчитанных согласно уравнению (6.82) и использованием указанных выше параметров значений температуры интенсивной деструкции для двух полимерных материалов приведено на рис. 6.63. Как можно видеть, получено хорошее соответствие теории и эксперимента (среднее расхождение величин ![]() и
и ![]() составляет ~ 2 %) [90].
составляет ~ 2 %) [90].

с = 1,25 ´ 10-3(Тc - 350 К). (6.83)
Уравнение (6.83) позволяет сделать ряд выводов. Во-первых, оно справедливо для полимеров с относительно высокой температурой стеклования (Тc > 350К). Во-вторых, увеличение Тc приводит к росту с и, согласно уравнению (6.82), к уменьшению роли структуры в определении Td. В третьих, последнее обстоятельство объясняет более высокие значения с для медленной диффузии по сравнению с быстрой для одного и того же полимера, роль структурного фактора в случае медленной диффузии существенно ниже, что можно было предположить априори. С учетом уравнения (6.83) формулу (6.82) можно переписать следующим образом [90]:
Df = 1,25´10-3(Tc-350К)(Td-Tc)b, (6.84)
что позволяет избежать разных значений коэффициента с для разных полимеров в случае быстрой диффузии.
На рис. 6.65 приведено сравнение экспериментальных ![]() и рассчитанных согласно уравнению (6.84)
и рассчитанных согласно уравнению (6.84) ![]() значений температуры интенсивной деструкции для углепластиков на основе фенилона, пленок ПК и стеклопластиков на основе ПАр. Как можно видеть, уравнение (6.84) дает достаточно точную оценку величины Td (среднее расхождение
значений температуры интенсивной деструкции для углепластиков на основе фенилона, пленок ПК и стеклопластиков на основе ПАр. Как можно видеть, уравнение (6.84) дает достаточно точную оценку величины Td (среднее расхождение ![]() и
и ![]() составляет ~ 2,8 %) в интервале Td ~ 160К [90].
составляет ~ 2,8 %) в интервале Td ~ 160К [90].
И наконец, на рис. 6.66 показана зависимость отношения (Td/Tc) от Тc, рассчитанная согласно уравнению (6.84). Как следует из графика рис. 6.66, отношение (Td/Tc) быстро стремится к единице или Td ® Tc по мере роста Тc и при Тc » 700 К различие между Тd и Тс находится в пределах погрешности расчета. Именно такое соотношение между Тd и Тс при повышении Тс наблюдается экспериментально [84].
Таблица 6.4
Сравнение экспериментальных и рассчитанных по уравнению (6.82) температур 20%-й потери массы для органопластиков на основе ПАр [92]
|
Армирующее волокно |
|
Тс, К |
Df |
b |
|
D, % |
|
|
Тип |
Содержание, масс. % |
||||||
|
Углен |
5 |
713 |
453 |
2,47 |
0,595 |
700 |
1,8 |
|
15 |
718 |
458 |
2,45 |
0,592 |
710 |
1,0 |
|
|
25 |
723 |
463 |
2,42 |
0,587 |
718 |
0,7 |
|
|
35 |
728 |
473 |
2,39 |
0,582 |
737 |
1,2 |
|
|
|
|
|
|
|
|
|
|
|
Терлон |
5 |
713 |
463 |
2,45 |
0,592 |
715 |
0,3 |
|
15 |
718 |
463 |
2,43 |
0,588 |
720 |
0,4 |
|
|
25 |
723 |
473 |
2,40 |
0,583 |
737 |
1,9 |
|
|
35 |
728 |
488 |
2,40 |
0,583 |
751 |
3,1 |
|
|
Вниивлон |
5 |
703 |
459 |
2,50 |
0,600 |
695 |
1,1 |
|
15 |
708 |
461 |
2,50 |
0,600 |
700 |
0,4 |
|
|
25 |
710 |
463 |
2,50 |
0,600 |
702 |
1,0 |
|
|
35 |
713 |
473 |
2,50 |
0,600 |
712 |
0,2 |
|
Авторы работы [92] использовали уравнение (6.82) для расчета температуры 20%-й потери массы образца в испытаниях ТГА в случае органопластиков на основе ПАр [80]. В табл. 6.4 приведено сравнение экспериментальных ![]() и теоретических
и теоретических ![]() значений Т20% для случая быстрой диффузии. Как можно видеть, получено хорошее соответствие теории и эксперимента (среднее расхождение D составляет ~ 1,1 %). Все рассчитанные значения
значений Т20% для случая быстрой диффузии. Как можно видеть, получено хорошее соответствие теории и эксперимента (среднее расхождение D составляет ~ 1,1 %). Все рассчитанные значения ![]() ложатся в интервал экспериментально определенных температур протекания термоокислительной деструкции - 673 ¸ 763К [80].
ложатся в интервал экспериментально определенных температур протекания термоокислительной деструкции - 673 ¸ 763К [80].
Характерно, что для этих органопластиков расчет ![]() для случая медленной диффузии также дал хорошее соответствие с экспериментом. Очевидно, это обусловлено значениями Df = 2,5 (табл. 6.4). Тем не менее, погрешность расчета в этом случае выше и составляет ~ 2,7% [92].
для случая медленной диффузии также дал хорошее соответствие с экспериментом. Очевидно, это обусловлено значениями Df = 2,5 (табл. 6.4). Тем не менее, погрешность расчета в этом случае выше и составляет ~ 2,7% [92].
Cледовательно, изложенные выше результаты показали важную роль структуры полимерного расплава в определении температуры интенсивной деструкции или термостойкости полимерных материалов и других характеристик ТГА. Термостойкость, как и температуры определенной потери массы образцов, зависит от химического строения полимеров, выраженного через температуру стеклования, компактности макромолекулярного клубка в расплаве и типа (интенсивности) диффузии оксиданта, который также определяется структурой клубка. Роль последней в определении термостойкости снижается по мере увеличения температуры стеклования полимера [90].