
Научная электронная библиотека
Монографии, изданные в издательстве Российской Академии Естествознания

Случаи синдрома Ретта вследствие микроделеций Xq28, затрагивающих ген МЕСР2
Несмотря на то, что интрагенные мутации гена MECP2 являются основной причиной RTT, существует небольшая часть случаев заболевания (5–10 %), когда MECP2-мутация не выявляется, и генетическая основа заболевания остается неясной. Ранее было описано несколько случаев микроделеций в участке Xq28, затрагивающих ген МЕСР2, у детей с фенотипическими проявлениями RTT [Ворсанова и др., 2013 в; Iourov et al., 2012]. Исходя из предположения о том, что субмикроскопические вариации числа копий последовательностей ДНК, затрагивающие целиком ген МЕСР2, могут быть нередкой причиной RTT, проведен их поиск с помощью технологии arrayCGH.
Как упоминалось ранее, когорта детей с RTT, у которых определялась MECP2 мутация традиционными молекулярно-генетическими
методами, насчитывала 354 ребенка: 262 случая классического и 92 случая атипичного RTT. Интрагенные мутации были определены в 96 % случаях классической формы и 71 % атипичных вариантов заболевания. 12 классических и 27 атипичных случаев заболевания, негативных по интрагенным МЕСР2-мутациям (39 девочек), были исследованы с использованием технологии arrayCGH. Удельный вес девочек с RTT, негативных по мутации гена МЕСР2 при молекулярно-генетическом тестировании, показан на рис. 44.

Рис. 44. Удельный вес девочек с синдромом Ретта, негативных по мутации гена МЕСР2 при молекулярно-генетическом тестировании
Было показано, что у 10 среди 39 обследованных девочек (пять классических случаев RTT и пять атипичных) выявлены микроделеции Xq28, захватывающие ген MECP2. Анализ полученных данных свидетельствует о том, что девочки имели микроделеции в участке q28 хромосомы Х, захватывающие целиком ген MECP2, а также прилегающие к нему последовательности ДНК за границами этого гена [Ворсанова и др, 2014а; Iourov et al., 2013]. Клиническими особенностями этих девочек являлось относительно легкое течение заболевания по сравнению с детьми, имеющими интрагенные MECP2 мутации. У больных с делециями наблюдалось более позднее начало заболевания, сохранный навык ходьбы, легкая диспраксия движений рук и отсутствие микроцефалии. Другими особенностями фенотипа были – относительно малая масса тела при рождении, а также наличие дополнительных клинических признаков – аномалий развития различных органов и микроаномалий (рис. 45, А, Г). Так у отдельных девочек наблюдались кожные симптомы – гемангиомы на коже спины и пигментные пятна в виде «брызг грязи» в области спины, напоминающие пигментные отложения при синдроме Блоха-Сульцбергера, ген которого также локализован в участке Xq28. У некоторых больных отмечались аномалии строения ЦНС на МРТ – гипоплазия червя мозжечка и кистозная трансформация эпидурального пространства в области грудного отдела позвоночника. Отмечались также поликистоз почек, персистирующее овальное окно и лицевые микроаномалии. У трех девочек с микроделециями были исследованы особенности Х-инактивации и обнаружен её неслучайный характер. Очевидно, что легкое течение заболевания у девочек с микроделециями было связано с феноменом сдвига Х-инактивации, вероятно, возникшим вследствие селективного преимущества клеток, в которых хромосома Х с делецией инактивирована. Ниже приведено клиническое описание одной из девочек с RTT, связанным с микроделецией Xq28.
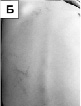
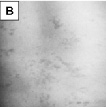
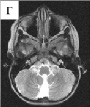
Рис. 45. Клинические особенности микроделеционного варианта синдрома Ретта: А – низкая масса тела при рождении; Б – гемангиомы на коже спины; В – пигментные пятна («брызги грязи») в области спины; Г – гипоплазия червя мозжечка на МРТ
Девочка родилась от молодых родителей (матери 24 года, отцу – 25 лет). Первая беременность матери закончилась спонтанным абортом на сроке 6 недель. Настоящая беременность – вторая, протекала с угрозой прерывания с 7-й до 20-й недели. Роды были физиологическими. Антропометрические показатели при рождении соответствовали нижней границе нормы (масса тела девочки – 2900 г, длина – 48 см, окружность головы – 34 см). Состояние при рождении расценивалось как удовлетворительное, а развитие – как соответствующее норме до возраста 10 месяцев, когда родители обратили внимание на задержку моторного развития (ребёнок не вставал самостоятельно). В возрасте 1 года девочка произносила 10 слов, однако, с 14 месяцев слова стали постепенно пропадать. Общение с родителями и сверстниками, интерес к играм и целенаправленные движения не были нарушены. Девочка начала ходить без поддержки с 16 месяцев, речевые навыки стали восстанавливаться с полутора лет. В последующие годы ребенок развивался с задержкой психоречевого развития. Генетиком по месту жительства были предположены и исключены на основании молекулярно-генетических исследований синдромы Ангельмана и RTT. При обследовании в возрасте 9 лет было обнаружено, что физическое развитие ребёнка было средним дисгармоничным с избытком массы тела. Отмечались следующие особенности фенотипа: выступающая переносица, короткая шея, гемангиомы, множественные пигментные пятна на коже спины (рис. 45, Б, В), длинные конусовидные пальцы кистей. Выявлялась умственная отсталость легкой степени. Периодически появляющиеся стереотипные движения рук напоминали характерные для RTT, поэтому была проведена оценка фенотипа ребёнка с помощью шкалы, разработанной нами для этого заболевания, которая показала низкий уровень суммарной оценки – 15 баллов. Обычно этот уровень наблюдался нами у детей со стёртой формой RTT. Относительная сохранность целенаправленных движений рук и речи, наличие на коже сосудистых пятен, противоречили диагнозу RTT. Однако, наличие у больной клинических признаков, характерных для этого заболевания, несмотря на их низкую экспрессивность, не позволяло окончательно его отвергнуть, даже при отсутствии мутаций гена MECP2, исследованных с помощью молекулярно-генетических методов.
При проведении серийной сравнительной геномной гибридизации у ребёнка обнаружена сложная аномалия генома в виде микроделеций хромосомы Х (участков Xq13.1, Xq22.1, Xq28) и микроделеции хромосомы 14 (участка 14q12). Кариотип пробанда после проведения arrayCGH записывался следующим образом: 46,XX,1phqh,arr14q12(23,575,187–23,732,133)x1,Xq13.1(70,474,715–70,608,113)x1,Xq22.1(100,082,182–100,164,698)x1,Xq28(153,145,800–153,301,421)x1. У девочки отмечалась делеция нескольких генов хромосомы 14 (участка 14q12), только один из которых связан с таким заболеванием, как болезнь специфических гранул нейтрофилов – один из видов иммунодефицита – ген CEBPE. Однако признаков иммунодефицита у девочки не выявлено. Был проведён анализ заболеваний, связанных с мутациями генов хромосомы Х, делеция которых была у девочки. Основным итогом проведенного анализа корреляции генотипа и фенотипа было выявление симптомов стёртой формы RTT и обнаружение делеции промотора и первых двух экзонов гена MECP2, мутации в котором вызывают заболевание. Ранее некоторыми авторами с помощью ряда молекулярно-генетических методов определялись крупные делеции, захватывающие несколько экзонов данного гена у тех больных с RTT, у которых не определялись мутации методом прямого секвенирования [Hardwick et al., 2007; Neul et al., 2008]. Очевидно, что у наблюдаемого нами ребёнка признаки RTT были связаны с потерей значительной части гена MECP2.
Таким образом, показано, что микроделеции, охватывающие участок хромосомы Х в области гена MECP2 (участок Xq28) и приводящие к полной делеции гена, этиологически и патогенетически могут быть связаны с RTT. Представленные в настоящей работе наблюдения демонстрируют преимущества использования высокоразрешающих молекулярно-цитогенетических технологий в диагностике Х-сцепленных форм умственной отсталости по сравнению с традиционными. Важно отметить, что без использования технологии молекулярного кариотипирования и оригинального биоинформатического метода оценки патогенности геномных перестроек, представленные в данной работе случаи были бы отнесены к идиопатическим формам умственной отсталости и аутистических расстройств [Ворсанова и др, 2014 б; Yurov et al., 2007 б; Vorsanova et al., 2010 а, б; Iourov et al., 2014]. Для обеспечения эффективности диагностики структурных аномалий хромосомы Х требуется использование комплекса методов: цитогенетического, молекулярно-цитогенетического (arrayCGH) и исследования инактивации хромосомы Х, которые позволяют выявлять ранее неизвестные формы заболеваний и определять медико-генетический прогноз.