
Научная электронная библиотека
Монографии, изданные в издательстве Российской Академии Естествознания

Cиндром микродупликации длинного плеча хромосомы Х, включающей ген MECP2
Наиболее часто выявляются дупликации, которые захватывают участок Xq28, включая ген MECP2. Так, в масштабных исследованиях вариаций генома при различных формах нарушения психики было показано, что среди геномных перестроек у мальчиков с недифференцированной умственной отсталостью 1 % составили дупликации длинного плеча хромосомы Х, включающие ген MECP2. В настоящее время в литературе описано более 130 индивидуумов из 36 семей с данной патологией [Lugtenberg et al., 2009; Sanlaville et al., 2009; Vissers et al., 2010; Neul, 2012; Van Esch, 2012; Shimada et al., 2013].
Дупликации Xq приводят к увеличению дозы генов терминального участка длинного плеча хромосомы Х, при этом наиболее часто наблюдается наличие дополнительного хромосомного материала в участке Xq28 [Breman et al., 2011], значительно реже отмечается перенос материала хромосомы Х в одну из аутосом, либо хромосому Y [Ворсанова и др., 2015]. Дистальные дупликации Xq большого размера, определяемые цитогенетическими методами, крайне редки (описано не более 50 случаев). Чаще встречаются дупликации малого размера, которые выявляются только с помощью высокоразрешающей технологии arrayCGH и, как правило, захватывают участок Xq28, включая ген MECP2 [Sanlaville et al, 2009]. Фенотип больных зависит от генного дисбаланса, вызванного дупликацией, наиболее значимым среди которых принято считать увеличение числа копий гена MECP2. Если дупликация включает данный ген, то формируется отличительный распознаваемый симптомокомплекс, описанный выше и носящий название синдрома дупликации гена MECP2, OMIM 300260 [Van Esch et al., 2005; Cheval et al., 2012]. При дистальных дупликациях Xq, не захватывающих ген MECP2, фенотип больных неспецифичен [Sanlaville et al., 2009]. Известно также, что с мутациями в гене MECP2 связан RTT (OMIM 312750). Дупликации гена MECP2 отличаются от известных интрагенных мутаций при RTT тем, что их результатом является не дефицит или аномальная структура соответствующего белка, а его гиперпродукция. Очевидно, что фенотип больных с дупликациями гена MECP2 отличен от такового при RTT.
Клинические проявления дистальных дупликаций Xq наиболее ярко проявляются у мальчиков. В первые недели жизни у пациентов наблюдаются выраженная диффузная гипотония мышц, нарушение глотания, гастроэзофагальный рефлюкс и избыточное слюнотечение, повышен риск аспирации. Дети плохо набирают массу тела. Типичны отставание в росте, микроцефалия, тяжелая задержка психического и моторного
развития. Если навык ходьбы развивается, то походка, как правило, неустойчива. У большинства больных отсутствует экспрессивная речь. Может наблюдаться регресс приобретенных двигательных и речевых навыков. Гипотония мышц постепенно сменяется спастичностью с формированием контрактур суставов. Примерно половина детей страдает эпилепсией, возраст манифестации которой широко варьирует. Наблюдаются миоклонии, абсансы, генерализованные тонико-клонические судороги, которые плохо контролируются антиконвульсантами. Отмечают специфические лицевые аномалии – брахицефалия, широкое лицо с полными щеками, эпикант, гипоплазия средней части лица, заостренный нос, маленький обычно полуоткрытый рот, крупные низко расположенные ротированные назад ушные раковины [Воинова и др., 2015; Chapleau et al., 2013; Na et al., 2013]. Больные страдают иммунодефицитом, что ведет к частым инфекциям верхних дыхательных путей, рекуррентным пневмониям с тяжелым течением, инфекциям среднего уха, синуситам. Известны единичные случаи менингита и инфекций уринарного тракта. Дети часто страдают запорами вследствие нарушений перистальтики кишечника. Наблюдаются признаки псевдообструкции кишечника, которые иногда при остром начале (со рвотой и болями в животе) расцениваются как «острый живот» и приводят к ургентной госпитализации. Отмечаются аномалии гениталий (гипоплазия, гипоспадия и крипторхизм), пороки сердца, маленькие кисти и стопы, аномалии пальцев (синдактилия, клинодактилия), снижение слуха, гипотиреоз. Большинство больных мужского пола умирают до достижения 25 лет [del Gaudio et al., 2006; Belligni et al., 2010; Petazzi et al., 2014]. Диагностика микродупликаций длинного плеча хромосомы Х стала возможной благодаря развитию высокоразрешающих полногеномных технологий, таких как array CGH [Ворсанова и др., 2013 б, в; Vorsanova et al., 2010 а, б;
Iourov et al., 2012].
Обследование родственников больных детей показало, что очень редко данная аномалия возникает de novo. В большинстве же случаев больные мальчики наследуют дистальную дупликацию длинного плеча хромосомы Х от своих матерей. Матери-носительницы часто не имеют
патологических клинических признаков благодаря выраженному сдвигу инактивации хромосомы Х, в результате которого в большинстве клеток женщин хромосома Х с дупликацией является неактивной. В некоторых случаях у лиц женского пола с дупликацией Xq, включающей ген MECP2, могут наблюдаться психические нарушения – тревожно депрессивные расстройства, специфические черты личности. Отдельными авторами сообщается о женщинах с преимущественной инактивацией нормальной хромосомы Х, у которых наблюдались тяжелые проявления заболевания: рекуррентные инфекции, недоразвитие речи, судороги [Fieremans et al., 2014; Novara et al., 2014]. Риск передачи микродупликации Xq матерью-носительницей ребенку составляет 50 %. Таким матерям при последующих беременностях рекомендуется пренатальная цитогенетическая и молекулярно-цитогенетическая диагностика [Ramocki et al., 2009]. Ниже представлены описания случаев дупликаций длинного плеча хромосомы Х, включавших ген MECP2, исследование которых проведено с помощью комплекса клинических и лабораторных методов. Помимо клинико-генеалогического метода использовалась рейтинговая шкала аутизма у детей CARS для оценки степени тяжести аутистических расстройств [Schopler et al., 1980]. Проводилось стандартное цитогенетическое исследование. Для молекулярного кариотипирования были использованы метафазная и серийная сравнительная геномная гибридизация на ДНК-микрочипах (CGH и arrayCGH) [Vorsanova et al., 2010 в; Iourov et al., 2012; 2014], содержащих 135 тыс. олигонуклеотидных проб, позволяющих сканировать геном с разрешением более 20 000 пн. Патогенность обнаруженных вариаций генома оценивали с использованием оригинальной биоинформатической технологии [Iourov et al., 2013; 2014].
Наблюдение 1. Пробанд – мальчик родился от молодых родителей (матери 26 лет, отцу – 29). Настоящая беременность была первой, протекала с угрозой прерывания в виде кровотечения на 14 неделе. В III триместре отмечалось многоводие. Роды произошли на 39 неделе, масса тела ребёнка при рождении была 3500 г, длина тела – 53 см. Мальчик приобрел двигательные навыки позже сверстников: голову держал с 4 месяцев, самостоятельно сидел – с 11 мес., ходил – с 16 мес., с полутора лет появились отдельные слоги. В возрасте полугода пробанд был консультирован неврологом, который отметил отсутствие зрительного контакта и задержку психомоторного развития (не следил за игрушкой, не переворачивался, не делал попыток сидеть). Ребенок часто страдал респираторными вирусными инфекциями (до 8 раз в год). Комплекс микроаномалий в 3,5 г. включал выступающий лоб, широкое лицо, фронтальный загиб волос вверх, эпикант, маленький нос с гипоплазией крыльев, маленький рот, крупные оттопыренные ушные раковины (рис. 43, А). Наблюдалась гипоплазия гениталий и шалевидная мошонка. В неврологическом статусе выявлялись гипотония мышц, выраженная задержка
психоречевого развития. Речь была представлена отдельными слогами. Выраженные нарушения поведения проявлялись отсутствием контакта с окружающими и интереса к игрушкам, приступами беспокойства. Оценка по шкале аутизма CARS (36 баллов) соответствовала умеренно выраженному аутизму. При ультразвуковом исследовании выявлено увеличение поджелудочной железы, при эхокардиографии – пролапс митрального клапана. Биохимические исследования основных показателей белкового, углеводного и липидного обмена, а также спектра аминокислот и ацилкарнитинов крови не выявили метаболических расстройств. На основании клинических проявлений ребенку был поставлен диагноз недифференцированная умственная отсталость, аутизм.
При цитогенетическом исследовании с применением дифференциального окрашивания хромосом по длине не было выявлено аномалий кариотипа. Исходя из клинических признаков, пациенту было проведено высокоразрешающее молекулярно-цитогенетическое исследование – метафазная сравнительная геномная гибридизация (CGH), позволяющая выявлять микроделеции и микродупликации по всему геному размером от 1,7 млн пн, что не позволяет сделать классическая цитогенетическая диагностика. После проведения CGH обнаружена микродупликация хромосомы Х в участке Xq28, которая после проведения CGH записывается в соответствии с международной классификацией как ish cgh dup(X)(q28qter).
У матери пробанда обнаружена неравная инактивация хромосомы Х (82:18), которая вместе со стёртыми клиническими признаками (когнитивные нарушения, широкое лицо, эпикант, маленький рот, крупные ушные раковины) может указывать на носительство ею микродупликации хромосомы Х. Наше наблюдение подтверждает мнение исследователей о том, что микродупликация хромосомы Х в участке Xq28, как правило, наследуется мальчиками от матерей, большинство которых имеет сдвиг Х-инактивации.
Наблюдение 2. Мальчик родился от четвертой беременности, наступившей у матери в возрасте 36 лет (отцу было 33 года). Мать имела здорового сына 12 лет от первого брака, две последующие беременности закончились медицинскими абортами. Отмечалась нефропатия в III триместре настоящей беременности. В связи с тазовым предлежанием плода на 39 неделе беременности проведено кесарево сечение: масса тела ребёнка была 4000 г, длина – 53 см. У новорожденного установлена дисплазия тазобедренных суставов, подвывих головки левого бедра, в связи с этим проводилась иммобилизация конечностей, после прекращения которой в возрасте 11 мес. возник рецидив вывихов головок обеих бедренных костей. В связи с иммобилизацией становление основных двигательных навыков на первом году жизни не отслеживалось, однако голову мальчик начал держать с задержкой (в 5–6 месяцев), и родители обратили внимание на отставание психоречевого развития ребёнка, словарный запас которого к году составлял 1–2 слова. Показатели физического развития, начиная со второго года жизни, оценивались как ниже среднего. С 1,5 лет появились приступы судорог в виде замирания с последующим тоническим
напряжением мышц конечностей и туловища, частота которых постепенно нарастала, к двум годам приступы стали ежедневными, а характер их сменился на генерализованные тонико-клонические судороги. Проводилась терапия антиконвульсантами. С появлением судорог произошёл некоторый регресс в психоэмоциональной сфере: появилось нарушение общения.
При обследовании в возрасте 4,5 лет физическое развитие мальчика было ниже среднего, гармоничное: длина тела – 99 см, масса тела – 15,5 кг. Выявлялись отдельные микроаномалии развития: широкая переносица, маленький рот, заострённый подбородок, крупные ушные раковины (рис. 43, Б); отмечалась нормальная окружность головы (51 см), нарушение контакта с ребенком, разнообразные стереотипные движения (раскачивание, хождение на носках по кругу) и другие аутистические черты в поведении. Оценка по шкале CARS соответствовала 32 баллам. Речевое развитие было на уровне использования 4–5 отдельных простых слов. Мальчик самостоятельно не ходил, наблюдались гипотония мышц лица и верхних конечностей, нижний вялый парапарез и тонико-клонические судороги в количестве 1–2 приступов ежедневно. На МРТ головного мозга выявлена дилатация желудочков мозга и умеренно выраженные признаки лиссэнцефалии. Клинический диагноз ребенка до получения результатов серийной сравнительной геномной гибридизации носил описательный характер: выраженная задержка психоречевого и моторного развития, симптоматическая эпилепсия, аутистические проявления.
При цитогенетическом анализе с применением дифференциального окрашивания хромосом по длине выявлен нормальный кариотип – 46,XY,1phqh. Учитывая тяжесть когнитивных нарушений и аномалию развития мозга на МРТ, было решено применить высокотехнологичный метод серийной сравнительной геномной гибридизации (arrayCGH) для исключения хромосомных микроаномалий. В результате проведенного исследования было обнаружено увеличение числа копий ДНК в длинном плече хромосомы Х, в участке Xq28. Результат исследования молекулярного кариотипа ребенка: arrXq28(153,130,000–153,647,227)x2. Дупликация составила примерно 500 000 пн.
Необходимо отметить, что у матери пробанда был обнаружен выраженный сдвиг Х-инактивации (96:4) при нормальном фенотипе, что может указывать на асимптоматическое носительство ею микродупликации Xq28. Данное наблюдение, как и предыдущие два, указывает на наличие в клинической картине у детей с микродупликациями Xq28 растройств аутистического спектра. Черты аутизма при этой патологии ранее не были описаны, возможно, из-за того, что психологический статус больных не исследовался. Анализ клинических признаков и выявление новых значимы для определения фенотипа, характерного для микродупликаций хромосомы Х в участке Xq28.
Наблюдение 3. Ребенок родился в срок от 1 беременности, протекавшей с угрозой прерывания на 24–26 неделе. Масса тела при рождении составила 2630 г., длина тела – 53 см, оценка по шкале Апгар – 7/8 баллов. В течение первых часов после рождения нарастали дыхательные расстройства: был переведен на искусственную вентиляцию легких на две недели. С рождения отмечалась задержка психомоторного развития: голову держал с 4 месяцев, не ползал, не ходил. Мальчик дважды перенес правостороннюю, верхнедолевую пневмонию в возрасте 4 и 11 месяцев. На МРТ головного мозга выявлены истончение мозолистого тела на всем его протяжении, выпячивание варолиевого моста, лобная атрофия без кортикального повреждения.
В возрасте 1 г. 7 мес. состояние ребенка расценено как тяжелое; физическое развитие было ниже среднего с массой тела 9,8 кг, длиной – 80 см. Окружность головы составляла 44 см и соответствовала выраженной микроцефалии. Комплекс микроаномалий включал брахицефалию, эпикант, широкое лицо, микростомию и открытый рот, низко расположенные крупные ушные раковины,
короткую шею (рис. 43, В); выявлены гипомимия лица, диффузная мышечная гипотония, гипорефлексия, отсутствие навыков самостоятельного сидения и ходьбы, импрессивной и экспрессивной речи, обеднение эмоциональной сферы, нарушение контакта с окружающими. Оценка по шкале CARS (41 балл) указывала на тяжелую степень аутизма. Отмечались хронические запоры, двусторонний крипторхизм. При исследовании методом arrayCGH, выявлена дупликация участка длинного плеча хромосомы Х – Xq27.3-q28, размером 10,6 Mb. Результат исследования
молекулярного кариотипа ребенка: arr Xq27.3q28 (144,313,065-154,930,046)х2. Размер дупликации у мальчика (более 10 млн пн) позволяет обнаружить подобную структурную аномалию цитогенетическим методом. Это исследование было проведено пробанду и родителям. Кариотипы родителей ребенка были нормальными, однако у отца ребенка была выявлена хромосомная нестабильность. Учитывая то, что большая часть матерей детей с дистальными дупликациями Xq могут быть бессимптомными носителями этой аномалии, матери пробанда было также проведено исследование arrayCGH, которое не обнаружило патологии. У пробанда при детальном анализе всех хромосом дополнительный участок был обнаружен не на хромосоме Х, как ожидалось, а на длинном плече хромосомы Y. Для уточнения локализации
дополнительного сегмента хромосомы X на хромосоме Y было проведено FISH исследование с ДНК зондом на ген MECP2, которое подтвердило его расположение на хромосоме Y. Таким образом, кариотип ребенка: 46,X,der(Y)t(X;Y)(q27.3;q12). Поскольку у отца ребенка хромосома Y была нормальной, можно предположить, что транслокация между хромосомами X и Y возникла в процессе мейоза у отца.
Последнее наблюдение представляет собой редкий случай микродупликации Xq28, возникшей de novo в результате транслокации участка хромосомы Х на хромосому Y. Проведенная комплексная генетическая диагностика способствовала корректному медико-генетическому консультированию данной семьи.
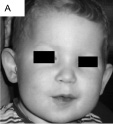

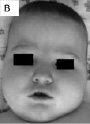
Рис. 43. Фенотипические особенности больных с микродупликациями длинного плеча хромосомы Х, участка Xq28, включающими ген MECP2: А – пробанд 1 (выступающий лоб, широкое лицо, фронтальный загиб волос вверх, эпикант, маленький нос с гипоплазией крыльев, маленький рот, крупные оттопыренные ушные раковины); Б – пробанд 2 (широкая переносица, маленький рот, заострённый подбородок);
В – пробанд 3 (высокий лоб, широкое лицо, эпикант, маленькие нос и рот)
Результаты сравнения фенотипов, наблюдавшихся нами у 3-х детей с дупликациями длинного плеча хромосомы Х, включающими ген MECP2, и описанных зарубежными авторами приведены в табл. 30.
Как видно из табл. 30, отмечался ряд общих для всех детей клинических признаков: микроцефалия, задержка психомоторного и речевого развития, гипотония мышц конечностей, судороги, нарушения вскармливания, гипоплазия гениталий, рекуррентные инфекции и лицевые микроаномалии (широкое лицо, эпикант, крупные ушные раковины, маленький рот). Следует отметить аутистические проявления у всех наблюдавшихся детей, которые не описывались ранее как характерный признак заболевания, возможно потому, что не были исследованы. На основании специфического фенотипа, большинство исследователей предлагают считать данное заболевание микродупликационным синдромом [Van Esch et al., 2005; Iourov et al., 2008; Sanlaville et al., 2009; Novara et al., 2014], что следует считать обоснованным, учитывая полученные нами клинические данные. Вместе с тем, отмечена вариабельность фенотипических проявлений синдрома. Некоторые признаки (отсутствие навыка ходьбы, гипотония лицевой мускулатуры, спастичность и др.) наблюдались не у всех, а только у одного либо двух больных. Отмеченные фенотипические различия между детьми, вероятно, зависели от размеров дупликации длинного плеча хромосомы Х в каждом конкретном случае.
Таблица 30
Сравнение клинических признаков у больных с дупликациями Xq28, включающими ген MECP2 из наших наблюдений 1, 2, 3, и литературы
|
Признаки, |
Случай № 1 |
Случай № 2 |
Случай № 3 |
Частота признака по данным литературы (количество больных с данным симптомом / количество клинических описаний) [Sanlaville et al., 2009] |
|
Задержка физического развития |
- |
- |
+ |
2/3 |
|
Микроцефалия |
- |
- |
+ |
5/39 |
|
Лицевые микроаномалии: |
||||
|
Эпикант |
+ |
- |
+ |
1/8 |
|
Крупные ушные раковины |
+ |
+ |
+ |
4/20 |
|
Маленький рот |
+ |
+ |
+ |
6/20 |
|
Широкое лицо |
+ |
- |
+ |
4/8 |
|
Неврологические симптомы: |
||||
|
Гипотония мышц конечностей |
+ |
+ |
+ |
29/32 |
|
Гипотония лицевой мускулатуры |
- |
- |
+ |
19/28 |
|
Задержка психомоторного развития |
+ |
+ |
+ |
47/47 |
|
Отсутствие речи или задержка речевого развития |
+ |
+ |
+ |
46/47 |
|
Отсутствие или ограничение ходьбы |
- |
- |
+ |
21/34 |
|
Спастичность |
- |
- |
- |
17/21 |
|
Судороги |
- |
+ |
+ |
22/42 |
|
Нарушения поведения (аутизм)* |
+ |
+ |
+ |
Не описаны |
|
Гипоплазия гениталий/крипторхизм |
+ |
- |
+ |
5/10 |
|
Проблемы со вскармливанием |
- |
- |
+ |
15/29 |
|
Хронические запоры |
- |
+ |
+ |
13/17 |
|
Аномалии пальцев |
- |
- |
- |
6/20 |
|
Рекуррентные инфекции |
+ |
+ |
+ |
33/40 |
Примечание: *Курсивом отмечен признак, наблюдавшийся у пробанда в нашей работе, но не отмеченный ранее у детей с дистальными дупликациями Xq.
Следует отметить, что состояние детей 1 и 2 с субмикроскопическими дупликациями Xq было более легким (сохранный навык ходьбы, отсутствие задержки физического развития, микроцефалии и проблемы со вскармливанием) по сравнению с наблюдаемыми у детей с дупликациями большого размера в наблюдении 3. Поскольку больные 2 и 3 были обследованы с помощью высокоразрешающей технологии array CGH, то был возможен детальный анализ корреляций генотипа и фенотипа у этих детей. Так, у больного 2 с относительно легкими клиническими признаками дупликация участка Хq28 составила всего около 500 000 пн по сравнению с дупликацией размером более 10 000 000 пн у больного 3 с тяжелым симптомокомплексом. Дуплицированнный участок в наблюдении 2 включал 26 генов, 7 из которых связаны с заболеваниями, описанными в базе данных OMIM. У больного 3 в общей сложности было дуплицировано 155 генов, 23 из которых ассоциированы с указанными в OMIM генетическими синдромами.
Принято считать, что к развитию патологии ЦНС и лицевых аномалий у больных с дистальными дупликациями Xq приводит повышенная экспрессия именно гена MECP2. Это послужило основанием назвать заболевание синдромом дупликации гена MECP2, несмотря на увеличение «дозы» других, обычно входящих в состав дупликации генов, таких как SLC6A8, L1CAM, FLNA, GDI1 и др. [Van Esch et al., 2005; Iourov et al., 2008б; Shimada et al., 2013]. Так, описаны наиболее тяжелые клинические проявления у больного с трипликацией гена MECP2 [Van Esch, 2012]. Кроме того, были описаны непатогенные дупликации Xq28, не включающие ген MECP2 [Lugtenberg et al., 2009]. Можно лишь частично согласиться с мнением этих авторов, поскольку некоторые факты свидетельствуют о том, что увеличение экспрессии других генов, помимо гена MECP2, вносят свой вклад в поражение ЦНС у больных с дупликациями длинного плеча хромосомы Х. Так, в отдельных исследованиях были обнаружены случаи увеличения числа копий участка Xq28, которые захватывали ген GDI1 (GDP dissociation inhibitor 1), но не затрагивали ген MECP2. При этом число копий гена GDI1 коррелировало с тяжестью нарушений интеллекта: от умеренной умственной отсталости у мальчика с дупликацией данного гена до тяжелого снижения интеллекта с эпилепсией и пороками головного мозга у двух братьев с пятью копиями гена GDI1 [Chapleau et al., 2013]. Кроме того, предполагается, что увеличение дозы гена GDI1 за счет дупликации длинного плеча хромосомы Х большого размера, может быть ассоциировано с наблюдаемой у части больных микроцефалией. Действительно, у больного 2 с отсутствием микроцефалии и меньшей тяжестью поражения ЦНС дупликация не захватывала ген GDI1, в то время как в случае 3 с выраженной микроцефалией и тяжелым поражением ЦНС этот ген был дуплицирован. Большинство исследователей считает, что иммунодефицит у больных связан с увеличением числа копий гена IRAK1 (interleukin-1 receptor-associated kinase 1), вовлеченного в развитие иммунной системы. Отмечено также, что если дупликация включает ген FLNA (Filamin A), то она обычно ассоциирована с нарушением перистальтики кишечника. Наши наблюдения согласуются с этими предположениями, поскольку у больных 2 и 3, у которых наблюдались частые инфекционные заболевания и хронические запоры, дупликация захватывала оба вышеупомянутых гена. Аномалии гениталий связывают с нарушением экспрессии гена MAMLD1 – Mastermind – like domain containing protein 1. В наблюдении 2 ген MAMLD1 не входил в состав дупликации, при этом нарушений строения гениталий не было отмечено. В то же время у пробанда 3 с крипторхизмом и гипоспадией дупликация захватывала данный ген. Полученные в нашем исследовании данные указывают на то, что увеличение дозы не только MECP2, но и других генов, вносит вклад в клинические проявления дупликаций длинного плеча хромосомы Х. Возможно, широкое применение термина «синдром дупликации гена MECP2» недостаточно корректно.
Согласно данным литературы, большая часть матерей детей, имеющих дупликацию участка Хq28, могут быть бессимптомными носительницами этой хромосомной аномалии, не проявляя клинических признаков заболевания за счет сдвига Х-инактивации, когда в организме женщины преимущественно инактивируется пораженная хромосома Х. В связи с предполагаемым высоким риском (50 %) повторного рождения больного ребенка мужского пола, в наблюдавшихся нами семьях было рекомендовано обследование родителей с последующим медико-генетическим консультированием. У матерей наших больных (матери детей в случаях 1 и 2) определен сдвиг инактивации хромосомы Х, на основании которого было предположено носительство женщинами из этих семей микродупликации длинного плеча хромосомы X, включающей ген MECP2. Им рекомендовано обследование методом array CGH, и, в случае выявления микродупликаций, направление на инвазивную пренатальную диагностику при последующих беременностях [Vissers et al., 2010]. В наблюдении 3 мать не являлась носительницей дупликации (по результатам arrayCGH), которая возникла de novo, на что указывало присутствие дополнительного материала хромосомы Х на хромосоме Y у пробанда и нормальная хромосома Y у его отца. В подобных случаях повторный риск рождения больного ребенка равен общепопуляционному.
Следует отметить, что синдром, связанный с микродупликациями длинного плеча хромосомы Х, включающими ген MECP2, был открыт в последние десятилетия благодаря развитию технологии arrayCGH [Van Esch et al., 2005]. Данная технология значительно повлияла на развитие медицины, увеличив возможности идентификации новых микроделеционных и микродупликационных синдромов, сопровождающихся поражением ЦНС [Ворсанова и др., 2013 б; Юров и др., 2013; 2015]. Открытие синдромов обычно основывается на исследовании корреляций фенотипа и генотипа больных. Исторически, выявление новых синдромов начиналось со всестороннего описания общих клинических признаков у однородной группы пациентов, после чего определялись связанные с данным симптомокомплексом генетические аномалии. Однако, в последние годы технология arrayCGH сделала доступным получение данных о генотипе больных. В результате на первый план при идентификации новых синдромов выступает анализ аналогичных геномных изменений в когорте детей, а затем определяются и сравниваются
их общие клинические признаки. Этот способ так называемого «обратного фенотипирования» [Юров и др., 2012; Ворсанова и др., 2014; Воинова и др., 2015] доказал свою эффективность, учитывая растущий список открываемых с его помощью новых синдромов, в который входит в том числе синдром микродупликации длинного плеча хромосомы Х, включающей ген MECP2.
Наши наблюдения демонстрируют то, что для обеспечения эффективной диагностики структурных аномалий хромосомы Х требуется использование нескольких методов: цитогенетического, молекулярно-цитогенетических (FISH, CGH, arrayCGH), а также исследования инактивации хромосомы Х. У каждого больного соответствие клинических признаков аномалиям, выявленным цитогенетическими и молекулярно-цитогенетическими методами, требует индивидуального анализа, который может способствовать поиску генов-кандидатов, изменение числа копий ДНК которых ведёт к формированию фенотипа. Комплексное обследование больных с аномалиями хромосомы Х и членов их семей, включающее все перечисленные выше методы, позволяет не только корректно проводить генетическую диагностику, но и определять медико-генетический прогноз.