
Научная электронная библиотека
Монографии, изданные в издательстве Российской Академии Естествознания

Исследование особенностей инактивации хромосомы Х в семьях с Х-сцепленной умственной отсталостью у детей
Нами был проведен анализ особенностей инактивации хромосомы Х у 247 индивидуумов женского пола из 150 семей с Х-сцепленными формами умственной отсталости, небольшой группы женщин (6 индивидуумов) из семей со структурными аномалиями хромосомы Х и 25 женщин из 22 семей с недифференцированными формами нарушений интеллекта. В контрольной группе исследовали 80 женщин без фенотипических проявлений врожденных и наследственных нарушений, а также не имеющих близких родственников с болезнями, связанными с аномалиями хромосомы Х. Применялся молекулярно-генетический метод, основанный на метил-чувствительной рестрикции участка Х-сцепленного гена андрогенового рецептора, включающего полиморфную последовательность тринуклеотидных повторов (ЦАГ)n, число которых в норме широко варьирует, что позволяет различить два аллеля гена андрогенового рецептора у лиц женского пола. Инактивация хромосомы Х определялась как соотношение инактивированных аллелей гена андрогенового рецептора разного родительского происхождения и оценивалась как неравная, если это соотношение превышало 20:80 (рис. 29, вверху). Чтобы избежать ошибок, связанных с неполной амплификацией и рестрикцией, учитывалась интенсивность аллелей в образцах ДНК, необработанных ферментом (HpaII-) (рис. 29, внизу). На рис. 30 представлены результаты количественного ПЦР-анализа инактивации хромосомы Х при случайной (равной, рис. 30, А) и неслучайной (неравной, рис. 30, Б) Х-инактивации.
Проводилось также исследование в парах «мать-пробанд», которое позволило установить направление сдвига инактивации хромосомы Х у женщин. На рис. 31 приведен пример анализа направления сдвига инактивации хромосомы Х: у женщины было определено два фрагмента ДНК разной длины, которые соответствуют двум аллелям гена AR, а у пробанда определен один фрагмент, который соответствует унаследованному от матери аллелю данного гена. Сопоставив их длины, можно установить, что в клетках женщины преимущественно инактивирована хромосома Х, унаследованная пробандом (т.е. несущая мутацию). У части женщин (N = 37) число CAG-повторов в обоих аллелях гена андрогенного рецептора было одинаково, как и в ранее проведенных исследованиях [Юров и др., 2005 б; Plenge et al., 2002]. В случаях, когда оба аллеля гена AR имели одинаковую длину, анализ Х-инактивации невозможно было провести. С учетом этих 37 случаев общая эффективность данного метода для всех образцов ДНК в настоящем исследовании составила 89,4 %, что соответствует данным литературы [Sharp et al., 2000].

Рис. 29. Результаты анализа инактивации хромосомы Х методом определения статуса метилирования экспансии (CAG)n повторов гена AR у женщины из семьи с Х-сцепленной умственной отсталостью. По оси абсцисс на электрофореграммах указана длина фрагментов ДНК (пн), по оси ординат – интенсивность флюоресцентного сигнала в относительных единицах флюоресценции – relevant fluorescent units (RFU). Электрофореграмма HpaII+ (вверху) соответствует амплифицированным участкам ДНК, обработанной метилчувствительной рестриктазой, а HpaII– (внизу) соответствует амплифицированным участкам необработанной ДНК. Стрелками выделены амплифицированные фрагменты гена AR. Пики одного и того же уровня показывают стандарт длины последовательностей ДНК


Рис. 30. Результаты анализа инактивации хромосомы Х: А – пример случайной (60:40), Б – неслучайной (91:9) Х-инактивации. HpaII+ соответствует ДНК, обработанной метилчувствительной рестриктазой, HpaII– соответствует необработанной ДНК. Стрелками выделены амплифицированные фрагменты гена AR
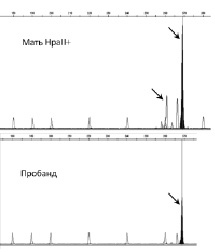
Рис. 31. Анализ направления сдвига инактивации хромосомы Х у матери пробанда с XLMR. У матери преимущественно инактивирована хромосома Х, которую унаследовал больной сын, т.е. хромосома Х с мутацией Х-сцепленного гена. Стрелками выделены амплифицированные фрагменты гена AR
Обследованные семьи с Х-сцепленными моногенными заболеваниями были разделены на 2 подгруппы. В первую входили Х-сцепленные формы умственной отсталости, проявляющиеся у мальчиков-гемизигот, при которых женщины-гетерозиготы имеют либо стертые признаки болезни, либо являются асимптоматическими носительницами (синдром умственной отсталости, сцепленной с ломкой хромосомой Х, синдромы Коффина-Лоури, Симпсона-Голаби-Бемеля, Барта, Лоу, гипогидротическая эктодермальная дисплазия и др.) В данной подгруппе Х-инактивация исследовалась у матерей, сестер и некоторых других родственниц. Во вторую подгруппу вошли семьи больных с Х-сцепленными синдромами, проявляющимися преимущественно у гетерозигот с внутриутробной летальностью для мальчиков-гемизигот. В данной подгруппе Х-инактивация исследовалась у пробандов женского пола и их матерей.
Результаты анализа инактивации хромосомы Х в исследованных группах семей показаны в табл. 19. В целом при Х-сцепленных синдромах, проявляющихся у девочек-гетерозигот, сдвиг Х-инактивации наблюдался у 45 % больных, а при Х-сцепленных заболеваниях, проявляющихся у гемизигот, сдвиг Х-инактивации определён у 46 % женщин из семей больных мальчиков (в 7 раз чаще, чем в контроле – 6,5 %).
Таблица 19
Удельный вес сдвига инактивации хромосомы Х у женщин из семей с Х-сцепленными формами умственной отсталости
|
Группы обследованных |
Удельный вес сдвига инактивации хромосомы Х среди индивидуумов женского пола |
Достоверность различий с контролем p* |
|
|
Абсолютное |
Относительное значение |
||
|
Женщины (матери и другие родственницы) из семей с Х-сцепленными формами умственной отсталости, проявляющимися преимущественно у гемизигот, в том числе: Синдром FRAXA; Синдром Хантера; Редкие синдромы Х-сцепленной умственной отсталости; Несиндромальная Х-сцепленная умственная отсталость |
30/67 8/19 2/15 15/26 5/7 |
45 % 42 % 13 % 58 % 71 % |
р < 0,001 |
|
Девочки с Х-сцепленными формами умственной отсталости, проявляющимися преимущественно у гетерозигот, в том числе синдромы: Блоха-Сульцбергера; Айкарди; Гольтца; Ретта |
38/82 10/10 1/1 1/1 26/70 |
46 % 100 % 100 % 100 % 37 % |
р < 0,001 |
|
Матери девочек с Х-сцепленными заболеваниями, проявляющимися преимущественно у гетерозигот, в том числе синдромы: Блоха-Сульцбергера; Айкарди; RTT |
13/66 1/5 0/1 12/60 |
20 % 20 % 0 % 20 % |
p < 0,01 |
|
Аномалии хромосомы Х |
5/5 |
100 % |
|
|
Женщины из семей с недифференцированными формами умственной отсталости |
6/23 |
26 % |
p < 0,01 |
|
Контрольная группа |
5/76 |
6,5 % |
|
Примечание. * – значение р оценивалось при сравнении каждой исследованной группы с контрольной группой методом дисперсионного анализа с использованием критерия Стьюдента.
Некоторые данные исследования Х-инактивации заслуживают подробного рассмотрения. Так, при обследовании 19 женщин из семей с синдромом умственной отсталости, сцепленной с ломкой хромосомой Х, у 8 из них обнаружен сдвиг Х-инактивации. Следует отметить преимущественную инактивацию хромосомы Х с экспансией тринуклеотидных повторов в гене FMR1 у 16 из 19 женщин. Только у 3-х женщин из 19 в клетках была преимущественно инактивирована нормальная хромосома Х. Полученные данные позволяют предположить, что экспансия тринуклеотидных повторов в гене FMR1 может вести к селекции клеток против тех, где активна хромосома Х с экспансией, что противоречит мнению некоторых исследователей, полагающих, что для заболеваний, связанных с увеличением числа CGG-повторов в гене FMR1 (синдромы FRAXA, POF), характерна случайная Х-инактивация [Migeon, 2007; Spath et al., 2010]. Согласно полученным нами данным о неслучайной инактивации хромосомы Х у женщин с экспансией CGG-повторов в гене FMR1 можно считать ее характерной для данной патологии, а преимущественная инактивация хромосомы Х с экспансией в клетках бόльшей части женщин указывает на существование при ней селекции клеток.
Особенности инактивации хромосомы Х были определены у 15 женщин-гетерозигот из семей с мукополисахаридозом II типа (МПСII, синдромом Хантера): 13ти матерей и 2-х сестёр пробандов. Лишь в двух случаях из 15 наблюдался сдвиг Х-инактивации, при этом у обеих женщин нормальная хромосома Х была активна в большинстве клеток. Все обследованные женщины не имели клинических проявлений МПСII. В литературе представлены единичные наблюдения за девочками с клиническими проявлениями синдрома Хантера, которые объясняются наличием у них сдвига инактивации хромосомы Х, способствующего экспрессии мутантного аллеля [Tuschl et al., 2005]. Подобные случаи, очевидно, чрезвычайно редки и нами не наблюдались. Направление сдвига Х-инактивации у обследованных женщин
из семей с МПСII было случайным, что согласуется с ранее проведенными
исследованиями, в которых показано отсутствие селекции клеток у гетерозигот и доказан факт метаболической кооперации клеток с активными мутантной и нормальной хромосомами Х [Migeon, 2007]. Полученные данные указывают на то, что Х-инактивация при синдроме Хантера носит случайный характер и является сбалансированной, по-видимому, вследствие отсутствия клеточной селекции. Таким образом, при синдроме Хантера исследования Х-инактивации не дают возможности выявления носительниц заболевания.
Х-инактивация была неслучайной у матерей детей с синдромами Коффина-Лоури, Барта, Симпсона-Голаби-Бемеля, FG и ото-палато-дигитальным синдромом 1 типа. При этом отмечена преимущественная инактивация той хромосомы Х, которая передана больному ребенку, что могло указывать на носительство женщинами Х-сцепленных мутаций. Таким образом, во многих семьях с редкими формами синдромальной Х-сцепленной умственной отсталости на основании анализа особенностей Х-инактивации выявлялись носительницы мутаций. Например, у двух сибсов с синдромом FG была определена одинаковая длина амплифицированого фрагмента ДНК гена AR, равная 268 пн (рис. 32).
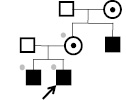
Рис. 32. Фрагмент родословной семьи с синдромом FG. Заштрихованы больные индивидуумы. Черной точкой внутри символов отмечены облигатные носительницы заболевания. Серой точкой вне символов отмечены доступные
для анализа индивидуумы
У матери сибсов со сдвигом Х-инактивации (87:13) в 87 % клеток была инактивирована хромосома Х с тем же аллелем гена AR (268 пн), который унаследован её больными сыновьями, т.е. хромосома Х с мутацией. Брат женщины был недоступен для исследования, однако по данным анамнеза имел признаки
синдрома FG.
Интересной иллюстрацией может служить наблюдение за семьей с Х-сцепленной спондилоэпифизарной дисплазией с умственной отсталостью (рис. 33, табл. 20).
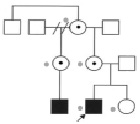
Рис. 33. Фрагмент родословной семьи с Х-сцепленной спондилоэпифизарной дисплазией с умственной отсталостью. В данной семье
для обследования были доступны 4 женщины и пробанд (отмечены серой точкой вне символов). Мать, бабушка и тетя пробанда были облигатными носительницами заболевания (отмечены чёрной точкой внутри символов)
У матери, бабушки и тети пробанда по материнской линии обнаружен сдвиг Х-инактивации (10:90, 17:83 и 11:89, соответственно), у сестры пробанда наблюдалась сбалансированная Х-инактивация (45:55). Фрагмент ДНК гена AR длиной 283 пн, обнаруженный у пробанда, определялся у его матери и бабушки и был инактивирован в большинстве их клеток. Сестра пробанда получила от матери другой аллель гена AR (277 пн). У тети пробанда (облигатной носительницы заболевания, имевшей сдвиг Х-инактивации) не определялось того аллеля гена AR, который унаследован пробандом (283 пн), что, по-видимому, произошло в результате рекомбинации генов хромосомы Х в половых клетках у бабушки пробанда. Следовательно, при анализе инактивации хромосомы Х необходимо учитывать возможность рекомбинации.
Таблица 20
Анализ Х-инактивации в семье с Х-сцепленной спондилоэпифизарной дисплазией с умственной отсталостью
|
Члены семьи |
Х-инактивация |
Размеры амплифицированных |
|
Бабушка пробанда |
17:83 |
280 и 283 пн |
|
Тетя пробанда |
11:89 |
259 и 280пн |
|
Мать пробанда |
10:90 |
277 и 283 пн |
|
Сестра пробанда |
45:55 |
265 и 277 пн |
|
Пробанд |
– |
283 |
При синдроме Коффина-Лоури и адренолейкодистрофии в клетках у матерей была обнаружена активная хромосома Х с мутацией, которая была передана пробанду. Известно, что при адренолейкодистрофии у гетерозигот с возрастом наблюдается селекция клеток в пользу тех, где активна хромосома Х, несущая мутацию, т.е. наблюдается «неблагоприятный» сдвиг Х-инактивации. Причина этого явления неясна, но необычное направление сдвига Х-инактивации объясняет появление ряда симптомов адренолейкодистрофии с возрастом у женщин-носительниц [Migeon, 2007]. Так, у 3 наблюдавшихся нами женщин из семей с адренолейкодистрофией хромосома Х с мутацией была активна в 58 %, 61 % и 71 % клеток. У самой старшей из них, 45-ти лет, наблюдались признаки заболевания в виде парестезий в области кистей и стоп. В клетках двух матерей детей с синдромом Коффина-Лоури также была преимущественно активна хромосома Х, несущая мутацию (рис. 34).

Рис. 34. Анализ Х-инактивации у матери ребенка с синдромом Коффина-Лоури. Сопоставление электрофореграмм матери и ребёнка показало, что у матери преимущественно инактивирована хромосома Х, которая не была передана пробанду. Стрелками выделены амплифицированные фрагменты гена AR

Рис. 35. Результаты исследования сдвига инактивации хромосомы Х у девочек с синдромом Блоха-Сульцбергера
У них наблюдалась неравная Х-инативация – 9:91 и 81:19 и имелись отдельные фенотипические черты заболевания: конической формы пальцы, периорбитальная полнота тканей, полные губы. Интеллект у обеих женщин был нормальным. Масштабные исследования Х-инактивации при синдроме Коффина-Лоури ранее не проводились. Следует отметить, что в литературе описаны женщины-гетерозиготы с синдромом Коффина-Лоури, имеющие легкие симптомы заболевания [Dobyns et al., 2006]. Нельзя исключить, что при данном синдроме (как и при адренолейкодистрофии) существует селекция клеток, способствующая экспрессии мутантного аллеля, что ведет к появлению клинических признаков у гетерозигот.
Таким образом, обнаружение сдвига Х-инактивации у женщин из семей мальчиков с XLMR указывало на носительство ими Х-сцепленных мутаций и высокий риск повторного рождения больного ребенка в семье. Эти данные были использованы нами при медико-генетическом консультировании.
Исследования Х-инактивации в семьях с моногенными заболеваниями, проявляющимися преимущественно у гетерозигот, позволили особо выделить синдром Блоха-Сульцбергера, при котором у всех наблюдавшихся нами больных (10 из 10 девочек) был обнаружен сдвиг Х-инактивации (рис. 35). Полученные результаты подтверждают высказывавшееся ранее мнение о том, что клетки с активной хромосомой Х, несущей мутацию гена IKBKG, подвергаются элиминации внутриутробно либо вскоре после рождения девочек с синдромом Блоха – Сульцбергера. При этом анализ Х-инактивации можно использовать как диагностический критерий данного заболевания, более того – его достаточно для подтверждения диагноза [Parrish et al., 1996]. В пяти семьях с синдромом Блоха-Сульцбергера, где для исследования были доступны родители, возможным было провести анализ происхождения инактивированной хромосомы Х (хромосомы Х с мутацией). Так, сопоставив длины амплифицированных фрагментов ДНК гена AR у ребенка и его родителей, было установлено, что у 3-х девочек хромосома Х с мутацией
имеет отцовское происхождение и у 2х – материнское. При отцовском происхождении хромосомы Х с мутацией делался вывод о ее возникновении de novo и родителям давался благоприятный прогноз. При материнском происхождении хромосомы Х с мутацией проводился анализ данных о характере Х-инактивации у матерей. Так, у матери близнецов с синдромом Блоха-Сульцбергера, несмотря на отсутствие клинических признаков заболевания, были основания предполагать возможное носительство мутации. У матери была определена случайная инактивация хромосомы Х, при этом в 61 % её клеток была активна хромосома Х, переданная обеим девочкам-близнецам. Наличие на этой хромосоме Х у матери мутации гена IKBKG было бы несовместимо с отсутствием симптомов заболевания. Следовательно, мутация, по-видимому, возникла de novo на хромосоме Х материнского происхождения. Существует, однако, небольшая вероятность того, что возникновение заболевания могло быть связано с герминативным мозаицизмом по мутации гена IKBKG у матери. Ранее это явление было описано при синдроме Блоха-Сульцбергера [Kirchman et al., 1995]. Это обстоятельство было учтено при медико-генетическом прогнозе. Материнское происхождение имела хромосома Х с мутацией еще у одной матери пробанда с синдромом Блоха-Сульцбергера. У этой женщины был обнаружен выраженный сдвиг Х-инактивации, при этом в большинстве клеток была инактивирована та же хромосома Х, что и у пробанда. У неё наблюдались депигментированные пятна на коже голеней и отсутствие 4-го верхнего зуба справа, т.е. стертые клинические признаки заболевания. Таким образом, исследование Х-инактивации позволило установить легкую форму синдрома Блоха-Сульцбергера у этой матери и информировать ее о высоком (50 %) риске рождения девочек с данной патологией. Таким образом, медико-генетическое консультирование семей с синдромом Блоха – Сульцбергера требует исследования Х-инактивации у матерей больных девочек и анализа происхождения хромосомы Х с мутацией.
У больной с фокальной дермальной гипоплазией обнаружен сдвиг Х-инактивации (99:1). Точковые мутации гена PORCN (porcupine homolog), вызывающего данное заболевание, обычно не ведут к сдвигу Х-инактивации, в то время как делеции данного гена вызывают сдвиг инактивации хромосомы Х [Leoyklang, 2008]. В настоящей работе мутации гена PORCN не были исследованы, однако, учитывая сдвиг Х-инактивации, можно предполагать, что девочка имела делецию данного гена.
В одной семье нами обследованы девочка с синдромом Айкарди и ее клинически здоровая мать. У пробанда определен сдвиг инактивации хромосомы Х (86:14), в то время как у матери Х-инактивация была случайной. У матери и ребенка в большинстве клеток была инактивирована одна и та же хромосома Х. Но отсутствие у матери клинических признаков при незначительном отклонении Х-инактивации позволило считать, что синдром Айкарди у ребенка – результат мутации de novo. Наше наблюдение согласуется с ранее проведенными работами, в которых сдвиг инактивации хромосомы Х обнаруживался у трети девочек с данной патологией и коррелировал с тяжестью неврологических симптомов [Eble et al., 2009].
Наибольшее количество обследованных семей с моногенными заболеваниями, проявляющимися преимущественно у гетерозигот, составили семьи с RTT. Среди девочек с данным заболеванием сдвиг Х-инактивации определен в 26 из 70 (37 %) случаев, а среди матерей в 12 из 60 (20 %). В нашей работе повышенный удельный вес неравной Х-инактивации при RTT совпадает с результатами, полученными многими авторами [Auranen et al., 2001; Weaving et al., 2003; Schanen et al., 2004; Bao et al., 2008]. В этих работах, как и в нашей, была преимущественно изучена классическая форма RTT. Можно сделать вывод о том, что феномен неравной инактивации хромосомы Х является характерной особенностью синдрома. Данные о зависимости фенотипических проявлений заболевания от особенностей Х-инактивации у детей с RTT представлены в разделе «Возможные корреляции генотипа и фенотипа при Х-сцепленных формах умственной отсталости и прогнозирование тяжести течения заболеваний».
Удельный вес сдвига Х-инактивации у матерей девочек с заболеваниями, сопровождающимися внутриутробной летальностью для гемизигот, составил 20 %. Это значение удельного веса ниже, чем у пробандов (46 %), и превышает контрольные значения более чем в 3 раза. В большинстве случаев заболевания с внутриутробной летальностью для гемизигот являются результатом мутаций de novo, поэтому низких значений удельного веса сдвига Х-инактивации у матерей следовало ожидать. Данные литературы об особенностях инактивации хромосомы Х у матерей девочек c заболеваниями, проявляющимися у гетерозигот, представлены лишь отдельными исследователями, которыми установлен сдвиг инактивации хромосомы Х у матерей больных с синдромами Айкарди (9 %) и RTT (20 %) [Юров 2005б; Bao et al., 2008; Eble et al., 2009].
Заслуживает внимания небольшая группа детей (6 пациентов) с аномалиями хромосомы Х, в семьях которых исследована Х-инактивация. Исследование Х-инактивации было возможно в 6 из 7 случаев: два мальчика с синдромом Клайнфельтера, одна девочка с делецией хромосомы Х и трёх матерей мальчиков, у которых обнаружена микродупликация хромосомы Х в участке Xq28. Так, у мальчика с регулярной формой синдрома Клайнфельтера (кариотип 47,XXY) определена неслучайная инактивация хромосомы Х (18:82) с преимущественной инактивацией аллеля гена AR бóльшей длины (длина аллелей гена AR – 271 и 274 пн). Это согласуется с наблюдениями авторов, которые обнаружили у трети больных с синдромом Клайнфельтера сдвиг Х-инактивации, что, по их мнению, может влиять на фенотипическую вариабельность заболевания [Iitsuka et al., 2001]. У другого ребёнка с регулярной формой синдрома Клайнфельтера длина двух аллелей гена андрогенового рецептора оказалась одинаковой (283 пн), что не позволило определить особенности Х-инактивации. Поскольку некоторыми авторами утверждалось, что клинические проявления синдрома Клайнфельтера (включая гинекомастию, остеопороз, нарушения социальной адаптации и когнитивные расстройства) тем тяжелее, чем больше количество CAG-повторов в гене AR [Migeon, 2007], нами проведено сравнение тяжести заболевания у двух обследованных больных. Полученные данные противоречат мнению других авторов, поскольку первый мальчик с меньшей длиной аллелей AR (271 и 274 пн) имел умственную отсталость, выраженные эмоционально-волевые нарушения, гипогенитализм и гинекомастию в отличие от второго мальчика (длина обоих аллелей гена AR – 283 пн), у которого наблюдались только пограничный интеллект и уменьшение размеров тестикул. У девочки с частичной делецией короткого плеча хромосомы Х (кариотип – 46,Х,del(Х)(р21р21) отмечен сдвиг Х-инактивации, равный 81:19. У ребенка наблюдались задержка психоречевого развития и аутизм. В силу недоступности родителей для исследования, происхождение преимущественно инактивированной хромосомы Х не определялось. Метод исследования не позволял установить, является ли она хромосомой Х с делецией. В ранее описанных подобных случаях структурных аномалий хромосомы Х была показана инактивация в большинстве клеток хромосомы Х с аномалией [Schluth et al., 2007].
У матерей двух мальчиков с микродупликациями Xq28, обнаруженными методом метафазной CGH (случай 1, табл. 30, представленный ниже) и arrayCGH (случай 2, табл. 30), был выявлен сдвиг инактивации хромосомы Х (82:18 и 96:4, соответственно). При этом в большинстве клеток каждой из матерей наблюдалась инактивация той хромосомы Х, которая унаследована сыном, т.е. хромосомы Х с дупликацией (рис 36), что позволяло с высокой вероятностью предположить носительство ими микродупликации хромосомы Х. У матери больного 3 наблюдался нормальный фенотип, что объяснялось выраженным сдвигом Х-инактивации, в то время как у матерей больных 1 и 2 определялись общие с ребенком признаки синдрома дупликации Xq28, такие как задержка речевого развития в анамнезе, легкой степени когнитивные нарушения, некоторые лицевые микроаномалии. Наличие общих с ребенком симптомов у этих женщин, вероятно, связано с тем, что в 18 % и 15 % их клеток, соответственно, была активной хромосома Х с дупликацией.
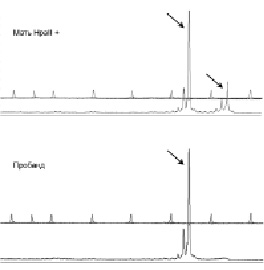
Рис. 36. Анализ инактивации хромосомы Х у матери пробанда с синдромом дупликации Xq28. На основании сопоставления длин фрагментов ДНК
гена AR, обнаруженных на электрофореграммах матери и пробанда, можно сделать вывод о том, что у женщины преимущественно инактивирована хромосома Х, унаследованная сыном. Стрелками выделены амплифицированные фрагменты гена AR
В последние годы в значительном числе работ, проводилось исследование инактивации хромосомы Х у матерей мальчиков со структурными аномалиями хромосомы Х. Практически во всех описанных случаях, когда матери являлись носительницами микроделеций или микродупликаций хромосомы Х, у них обнаруживалась неслучайная Х-инактивация [Van Esch et al., 2005; del Gaudio et al., 2006; Friez et al., 2006; Froyen et al., 2007; Madrigal et al., 2007; Bauters et al., 2008б; Gecz et al., 2009]. У всех этих женщин наблюдалась преимущественная инактивация хромосомы Х с аномалией и отсутствие патологических клинических признаков. В упомянутых работах, однако, не приводится детального клинического описания фенотипа матерей, и у них могли быть не зарегистрированы стертые клинические признаки. Сдвиг Х-инактивации, таким образом, может служить критерием отбора больных для поиска структурных аномалий хромосомы Х, как указывалось ранее некоторыми исследователями [Madrigal et al., 2007; Bauters et al., 2008 б].
Наиболее значимым, на наш взгляд, являлся анализ Х-инактивации у женщин в семьях с недифференцированными формами умственной отсталости: у 6 из 23 женщин был выявлен сдвиг Х-инактивации, при этом в большинстве клеток была инактивирована хромосома Х, переданная пробанду, что высоковероятно указывало на Х-сцепленный характер наследования патологии. Таким образом, почти в четверти случаев умственной отсталости, относившейся ранее к недифференцированной, можно было предполагать ее Х-сцепленный характер на основании сдвига Х-инактивации у матерей. Использование данного критерия при недифференцированной умственной отсталости имеет исключительно большое значение, поскольку этиология 25–45 % случаев тяжелой и большинства случаев легкой умственной отсталости остается невыясненной [Chelly, Mandel, 2001]. Обращает на себя внимание, что полученная величина 6 из 23 (условно 26 %) довольно близка к теоретически рассчитанной нами при определении удельного веса XLMR среди других форм нарушений интеллекта на основе соотношения полов (28,5 %). Напомним, что клинико-генеалогический метод позволил нам выявить XLMR только у 6,54 % больных с когнитивными нарушениями. Следовательно, анализ Х-инактивации позволяет обнаружить те случаи XLMR, которые оставались не выявленными после проведения клинико-генеалогического анализа. Полученные нами данные суммированы на рис. 37. Необходимо отметить, что 5/76 (6,5 %) женщин контрольной группы также имели сдвиг Х-инактивации. Последних, по-видимому, следует отнести в группу риска носительства Х-сцепленных генетических дефектов.
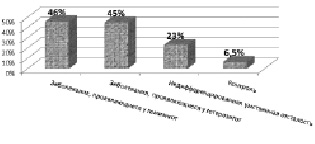
Рис. 37. Частота сдвига Х-инактивации при Х-сцепленных и недиференцированных формах умственной отсталости в сравнении с контролем
Полученные нами данные о высокой частоте сдвига инактивации хромосомы Х у женщин из семей с Х-сцепленной умственной отсталостью близки к данным других исследователей. Так, работы, посвященные сдвигу Х-инактивации у девочек с Х-сцепленными формами умственной отсталости, позволили установить ее сдвиг от 28 % до 37 % случаев при RTT, в 33 % случаев при синдроме Айкарди и 98 % случаев синдрома Блоха-Сульцбергера [Юров и др., 2005б; Parrish et al., 1996; Weaving et al., 2003; Bao et al., 2008; Eble et al., 2009]. Однако все проведенные ранее исследования были посвящены отдельным нозологическим формам, за исключением работы Plenge с соавторами [2002], в которой данные о частоте сдвига Х-инактивации (47 %) у женщин из семей мальчиков с различными формами XLMR, совпадают с полученным нами значением (45 %). В указанной работе исследовалось значительно меньшее число женщин (94), чем в нашей (250), и все они были только из семей с моногенными формами XLMR, проявляющимися у мальчиков-гемизигот, в то время как у нас значительную группу составили девочки с формами XLMR, проявляющимися у гетерозигот, и их матери. Нами были обследованы также индивидуумы из семей с аномалиями хромосомы Х и семей с недифференцированной умственной отсталостью. Наконец, исследованные Plenge формы XLMR не совпадали с изученными в нашей работе. Итак, исследования Plenge с соавторами и проведенное нами дополняют друг друга. Вместе они позволяют судить о том, что около половины индивидуумов женского пола из семей больных с XLMR являются носительницами Х-сцепленных мутаций.
Таким образом, неслучайная инактивация хромосомы Х является одной из характерных эпигенетических особенностей Х-сцепленных форм умственной отсталости (Юров и др., 2004б; 2005б; Plenge et al., 2002). Применение исследований Х-инактивации даёт возможность с высокой вероятностью предположить диагноз XLMR, выявить женщин-носительниц данных заболеваний и повысить эффективность медико-генетического консультирования в таких семьях.