
Научная электронная библиотека
Монографии, изданные в издательстве Российской Академии Естествознания

Фенотипическая характеристика редких Х-сцепленных синдромов умственной отсталости (синдромы Симпсона-Голаби-Бемеля, антера, Блоха-Сульцбергера, FG, ото-палато-дигитальный, фокальная дермальная гипоплазия Гольтца)
Синдром Симпсона-Голаби-Бемеля I типа проявляется, как правило, у мальчиков и в большинстве случаев связан с мутациями гена GPC3, кодирующего белок глипикан-3, который функционирует как рецептор на поверхности клеток, позволяющий регулировать клеточный ответ на факторы роста. Мутации в гене GPC3 приводят к нарушению регуляции глипиканом-3 клеточного ответа на факторы роста, что ведет к увеличению пролиферативной активности клеток, предрасположенности к возникновению новообразований, формированию фенотипа синдрома Симпсона-Голаби-Бемеля I типа [Budny et al., 2006]. Первые признаки синдрома можно предположить еще внутриутробно, начиная со срока беременности 16 недель. Наблюдаются многоводие, макросомия, висцеромегалия, краниофациальные аномалии, аномалии почек, полидактилия, диафрагмальные грыжи при ультразвуковом исследовании плода. Масса тела новорожденных может достигать 6 кг и более. В периоде новорожденности могут наблюдаться угрожаемые для жизни состояния – гипогликемия и обструкция дыхательных путей. В дальнейшем показатели длины, массы тела и окружности головы превышают возрастную норму на 2–3 стандартных отклонения [Sakazume et al., 2007]. Основными фенотипическими признаками синдрома являются макроцефалия и характерные особенности лица: грубые черты, антимонголоидный разрез глазных щелей с гипертелоризмом, эпикант, короткий широкий нос с широкой плоской переносицей и вздернутым кончиком, большой рот, макроглоссия, короткая уздечка языка, высокое небо, срединная бороздка на нижней губе, широкая выступающая нижняя челюсть. Общий вид пациентов – «бульдогообразный», в связи с чем одним из синонимов названия синдрома Симпсона-Голаби-Бемеля является «синдром бульдога». У больных с синдромом Симпсона-Голаби-Бемеля I типа, как правило, встречаются аномалии развития опорно-двигательного аппарата, включающие синдактилию II и III пальцев кистей и стоп, полидактилию, широкие I пальцы кистей и стоп, синостозы II-III шейных позвонков, добавочные ребра, добавочный поясничный позвонок, аномалии крестцового и копчикового отделов позвоночника, воронкообразную деформацию грудной клетки. К более редким симптомам относят расщелины верхней губы и неба, пороки развития сердечно-сосудистой, мочевыделительной, центральной нервной систем и желудочно-кишечного тракта. Среди аномалий мочеполовой системы встречаются нефромегалия, поликистоз почек, удвоение почечной лоханки, дольчатые почки, гипоспадия и др. Синдром Симпсона-Голаби-Бемеля относится к группе заболеваний с предрасположенностью к развитию новообразований. Частота развития опухолей составляет 10 %, большинство их них (94 %) локализованы в абдоминальной области и являются злокачественными. Наиболее часто регистрируются опухоль Вильмса, гепатобластома, нейробластома надпочечника, гонадобластома и гепатоцеллюлярная карцинома [Waterson et al., 2010]. На рис. 7 представлен фенотип сибсов с синдромом Симпсона-Голаби-Бемеля I типа в возрасте 15 и 16 лет.


Рис. 7. Фенотип сибсов с синдромом Симпсона-Голаби-Бемеля I типа: грубые черты лица, короткий широкий нос с широкой плоской переносицей,
большой рот, срединная бороздка на нижней губе, широкая выступающая нижняя челюсть
У братьев отмечались очень высокие показатели длины и массы тела, превышавшие 97 перцентиль. Наряду с этим наблюдались макроцефалия, грубые черты лица, прогнатия, синофриз, крупные кисти и стопы, поперечные складки на ладонях. Психическое развитие мальчиков соответствовало легкой степени умственной отсталости. У них выявлена дилатация левого желудочка, у старшего сибса – увеличение размеров селезенки, у младшего – увеличение размеров почек. У младшего брата отмечалась врожденная воронкообразная деформация грудной клетки II степени, не наблюдавшаяся у старшего. У матери – носительницы заболевания имелись следующие клинические признаки: высокие антропометрические параметры, наличие характерных грубых черт лица, брахидактилия кистей и стоп. У братьев и их матери определена мутация c.1159C>T (R387X) в экзоне 5 гена GPC3, что позволило подтвердить клинический диагноз синдрома Симпсона-Голаби-Бемеля I типа.
Синдром FG (синоним – синдром Опица-Каведжиа, OMIM 305450) упомянут выше в качестве примера явления синдромального расщепления в связи с выраженной генетической гетерогенностью. Данный фенотип наблюдается преимущественно у мальчиков при мутациях ряда Х-сцепленных генов: MED12 (наиболее часто), FLNA, CASK, UPF3B. Типичны задержка роста, относительная макроцефалия, высокий выступающий лоб, фронтальный загиб волос вверх, гипертелоризм, опущенные наружные углы глазных щелей, широкие первые пальцы кистей и стоп, сенсоневральная тугоухость, запоры. Возможны обструкция анального отверстия, крипторхизм, пороки сердца (аортальный стеноз). Признаками поражения ЦНС при заболевании являются умственная отсталость, судороги, гипотония, особенности поведения в виде дружелюбия, любопытства и гиперактивности с дефицитом внимания, полная или частичная агенезия corpus callosum на МРТ [Opitz et al., 2008]. Нами наблюдался семейный случай синдрома FG у двух сибсов 3 и 5 лет, которые отставали в физическом и психоречевом развитии с первых месяцев жизни, у старшего брата до 2-х лет отмечались частые респираторно-аффективные приступы, а с 3-летнего возраста кратковременные тонические судороги с частотой 1–2 раза в год. При поступлении в клинику оба ребенка имели задержку роста, относительную макроцефалию, долихоцефальную форму черепа с выступающим лбом, легкой степени умственную отсталость, гиперактивность и особенности личности в виде повышенной общительности и приветливости, гипотонию мышц, хронические запоры и характерный комплекс микроаномалий (рис. 8). Мать сибсов была низкого роста (153 см), с 9-летнего возраста страдала эпилепсией, периодически – депрессией, тяжелыми запорами и имела некоторые общие с детьми микроаномалии – высокий лоб и седловидный нос. Интеллект у матери был сохранен. Дядя сибсов по материнской линии был невыского роста (159 см), наблюдался психиатром по поводу легкой умственной отсталости, страдал запорами и имел такие внешние признаки, как относительная макроцефалия, выступающий лоб, опущенные наружные углы глаз, которые позволили предполагать у него синдром FG.
Сравнение клинических признаков у членов семьи позволило обнаружить ряд различий. У старшего сибса и матери, в отличие от других больных родственников, наблюдались судороги, а у младшего была более выражена умственная отсталость, у дяди отмечались приступы агрессивного поведения. Таким образом, синдром FG отличался
внутрисемейным клиническим полиморфизмом. Один из Х-сцепленных генов, мутации которого встречаются при синдроме FG, а именно ген фламина А (FLNA), связан с несколькими фенотипами, являясь примером синдромального смешивания (syndrome lumping) (табл. 6).
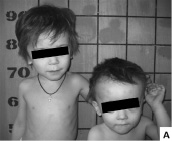
Рис. 8. Особенности фенотипа у детей 3 и 5 лет с синдромом FG: А – низкий рост, макроцефалия, высокий лоб; Б – выступающий лоб, седловидный нос; В – широкие большие пальцы и частичная синдактилия II–III пальцев стоп
К редким Х-сцепленными синдромами с умственной отсталостью относится ото-палато-дигитальный синдром 1-го и 2-го типа (OPD1 и OPD2, OMIM 311300 и 304120), который является следствием мутаций в гене FLNA [Fitch et al., 1983; Robertson et al., 2001]. Фенотип ото-палато-дигитального синдрома 1-го типа включает сочетание низкого роста, интеллектуального дефицита, кондуктивной тугоухости вследствие аномалий слуховых косточек, расщелины нёба, аномалий лица (плоское лицо, гипертелоризм, маленькие нос и рот с опущенными углами, скошенный лоб, отсутствующие и непрорезавшиеся зубы) и умеренно выраженной патологии скелета (отсутствие фронтальных и сфеноидальных пазух, деформации пальцев, грудной клетки, сколиоз). Фенотип ото-палато-дигитального синдрома 2-го типа более тяжелый и включает помимо задержки роста глубокую умственную отсталость, тяжелые аномалии скелета, сочетание различных пороков мозга, сердца, кишечника и почек, которые часто ведут к гибели больных детей в перинатальном периоде. Также как и при OPD1, выявляют кондуктивную тугоухость и расщелины нёба. Аномалии лица при OPD2 выражены в бόльшей степени (выступающий лоб, низко посаженные аномальные ушные раковины, гипертелоризм, маленький рот, маленькая нижняя челюсть).
У наблюдавшихся нами детей из разных семей с OPD1 и OPD2 клинические симптомы значительно различались по своей тяжести. Ребенок с синдромом OPD1 родился на 39 неделе беременности, протекавшей с угрозой прерывания и внутриутробной гипотрофией 2–3 степени, угнетением ЦНС, низкой массой (2350 г) и длиной (47 см) тела. Мальчик развивался с умеренной задержкой физического и психомоторного развития (сидит с 7 мес., ходит с 15 мес., говорит фразами с 3х лет). Ребенок с синдромом OPD2 родился недоношенным на 35 неделе беременности в тяжелом состоянии с низкой массой тела 1390 г. Гестационный возраст плода был оценен неонатологом как 30–32 недели. При анализе клинических проявлений у детей с синдромами OPD1 и OPD2 обращала на себя внимание значительная тяжесть течения заболевания во втором случае, которая привела к смерти ребенка в возрасте 1 года 9 месяцев. У ребенка с синдромом OPD2 отмечались значительно более тяжелые аномалии скелета, глубокая умственная отсталость, выраженные тугоухость и расщелина нёба. Кроме того у него наблюдались аномалии мозга (в виде расширения желудочков и полости прозрачной перегородки на МРТ) и сердца (открытое овальное окно), которые не выявлялись у ребенка с OPD1. Значительно отличались и особенности лица (рис. 9). У больного с OPD2 комплекс лицевых микроаномалий включал выступающий лоб, гипертелоризм, запавшую переносицу, маленькую нижнюю челюсть – признаки, которые не выявлялись у первого ребенка.
Выраженные отличия в клинических проявлениях синдромов OPD1 и OPD2 не вызывали сомнений. Это обстоятельство лежало в основе разделения синдромов OPD на две нозологические формы до открытия их единой этиологии. Согласно нашим наблюдениям при синдромах OPD1 и OPD2 клиническая картина включает патологию одних и тех же органов и систем, но разной степени тяжести (умственная отсталость, срединные расщелины, тугоухость и разнообразные аномалии скелета, включая деформации пальцев). Можно согласиться с мнением отдельных исследователей, которые считают, что описанные заболевания представляют собой единую нозологическую форму [Verloes et al., 2000].

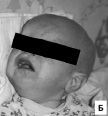
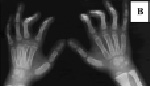
Рис. 9. Особенности фенотипа у детей с ото-палато-дигитальными синдромами 1-го и 2-го типов: А – эпикант, выступающие надбровные дуги у ребенка с синдромом OPD1; Б – выступающий лоб, маленькая нижняя челюсть у больного с синдромом OPD2; В – деформации пальцев
на рентгенограмме кистей у ребенка с синдромом OPD1
В основе мукополисахаридоза II типа (МПСII, синдрома Хантера) лежат мутации Х-сцепленного гена идуронатсульфатазы, кодирующего одноимённый лизосомный фермент. В результате недостаточной активности идуронатсульфатазы, участвующей в первом этапе катаболизма гликозаминогликанов (ГАГ) – гепаран- и дерматансульфата, возникает их аккумуляция в лизосомах практически всех типов клеток различных тканей и органов [Wraith et al., 2008]. МПСII страдают, как правило, мальчики. В типичных случаях заболевание характеризуется манифестацией на первом году жизни, прогрессирующим снижением интеллекта, появлением грубых черт лица, скелетными деформациями, гепатоспленомегалией. Характерно узелково-папуллезное поражение кожи, преимущественно в области лопаток, наружных и боковых поверхностей плеч и бедер, обусловленное отложением липидов и гликозаминогликанов. Макроглоссия, сужение глоточного кольца, трахеи и бронхов вследствие отложения ГАГ ведет к дыхательной обструкции. Другими симптомами заболевания являются кардиомиопатия и аномалии клапанов сердца, пупочные и паховые грыжи, снижение слуха, апноэ во время сна. Различают легкую и тяжёлую формы синдрома, обусловленные разными мутациями в гене идуронатсульфатазы. Больные с тяжелой формой имеют значительное нарушение интеллекта и погибают в конце второй декады жизни, а дети с более легкой формой заболевания отличаются нормальным интеллектом, продолжительность их жизни может составлять 50–60 лет. У больных наблюдается повышенная экскреция гепаран- и дерматансульфата, а также значительное снижение активности фермента идуронатсульфатазы, определяемого в лейкоцитах, фибробластах или плазме крови. Знание клинических проявлений этого синдрома приобрело особое значение в связи с разработкой патогенетической терапии с помощью генно-инженерного фермента идурсульфазы, которая позволяет корректировать соматические проявления заболевания [Семячкина и др., 2007]. У детей с МПСII (мы наблюдали 18 больных) были типичны грубые черты лица: утолщение губ, ноздрей, увеличение языка. Волосы были жесткими, сухими и лишенными блеска, отмечался гирсутизм. У всех детей наблюдались, контрактуры суставов, увеличение печени и селезенки, поражение клапанов сердца, частые рецидивирующие отиты и риниты. Характерными были шумное дыхание и низкий грубый голос. Особенности фенотипа детей с МПСII представлены на рис. 10. Перечисленные выше признаки наблюдались в 100 % случаев, несмотря на то, что дети значительно различались по возрасту: 5 детей обследовались в возрасте от 2 до 5 лет, 7 больных – от 6 до 10 лет, 4 ребенка – от 10 до 14 лет и 2 больных – старше 15 лет. Во всех случаях диагноз был лабораторно подтвержден при исследовании активности идуронатсульфатазы в лимфоцитах, которая была крайне низкой – от 0,01 до 2,4 нМ/мг/24 часа (при норме 18,8 – 82 нМ/мг/24часа). Вместе с тем у больных отмечался полиморфизм клинических проявлений. Так, возраст появления первых признаков заболевания (грубых черт лица, контрактур суставов) варьировал от года до 4 лет. Некоторые признаки наблюдались не во всех случаях и были различной степени выраженности. Отставание в росте наблюдалось у половины больных, макроцефалия – в 14 случаях. Снижение интеллекта отмечалось у 15 из 18 детей, и было различной степени тяжести. Другие признаки поражения нервной системы наблюдались реже: спастический тетрапарез – в 3 случаях, судорожный синдром – у 3 больных, выраженное агрессивное поведение – у 4 детей. Снижение слуха обнаруживалось у 7 больных.
При проведении эхокардиографии у 5 из 18 обследованных детей с МПСII выявлялась гипертрофическая кардиомиопатия. Хотя поражение клапанов сердца наблюдалось во всех случаях, характер его различался по степени тяжести: у 11 детей отмечались утолщение и ригидность клапанов, в 5 случаях сформировалась недостаточность митрального либо аортального клапанов и в 2 случаях – аортальный стеноз. Помимо гепатоспленомегалии при ультразвуковом исследовании паренхиматозных органов у 6 больных обнаружено увеличение размеров почек и поджелудочной железы, у 4 детей – только почек, у 3 – только поджелудочной железы. Грыжи наблюдались в 13 случаях: у 8 детей – одновременно паховые и пупочная, у 1 – пупочная и белой линии живота, у 2 больных – только паховые и у 2 – только пупочная грыжа. Типичные для МПСII узелковые изменения кожи отмечены у 3 больных.
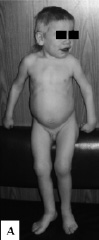
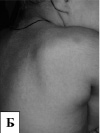
Рис. 10. Фенотип детей с мукополисахаридозом II типа: А – грубые черты лица, короткая шея, контрактуры суставов, увеличение живота за счет гепатоспленомегалии; Б – узелковые изменения кожи в области спины и гирсутизм
Таким образом, несмотря на сходство симптомов у больных с МПСII, наблюдались различия в частоте и экспрессивности многих фенотипических признаков.
Синдром Блоха-Сульцбергера (синдром недержания пигмента, inconinentia pigmenti, OMIM 308300) связан с мутациями в гене IKBKG [Fusco et al., 2004]. Заболевание характеризуется эмбриональной летальностью для гемизигот, поэтому его можно наблюдать только у девочек-гетерозигот (за редким исключением мальчиков с кариотипом 47,XXY). Ген IKBKG кодирует основной белок-модулятор нуклеарного фактора kappaB (NF-kappaB essential modulator protein). Дефицит данного белка ведет к апоптозу. Клетки с мутациями гена IKBKG на активной хромосоме Х становятся чрезвычайно подверженными апоптозу, что объясняет эмбриональную летальность мальчиков с IKBKG-мутациями. У девочек-гетерозигот внутриутробно в различных тканях происходит гибель клеток с мутацией гена IKBKG на активной хромосоме Х. Гибель клеток кожи с мутантным аллелем на активной хромосоме Х наблюдается у девочек сразу после рождения [Ardelean, Pope, 2006], что выявляется в виде поражений кожи в неонатальном периоде. Изменения кожи характеризуются стадийностью: эритематозно-везикулезная сыпь в первые 2 недели жизни, бородавчатые высыпания в течение нескольких лет, пигментные пятна в виде брызг грязи и далее мраморный рисунок пигментации кожи. Кроме того, наблюдаются аномалии волос, ногтей, зубов, глаз (косоглазие, катаракта), умственная отсталость, судороги [Landy, Donnai, 1993].
Нами обследованы 13 девочек из 12 семей с синдромом Блоха-Сульцбергера в возрасте от 2 недель до 15 лет. Среди них был один случай заболевания у близнецов, которые родились на 32-й неделе беременности с массой тела 1868 и 1750 г и длиной – 42 см в тяжелом состоянии с синдромом угнетения ЦНС. Остальные дети родились в срок и их состояние при рождении расценивалось как удовлетворительное, средние масса тела составила 3080 г (от 2750 до 3430 г), длина – 50,5 см (от 49 до 51 см). Манифестация заболевания наблюдалась с первых по пятые сутки жизни в виде появления эритемы, мелкопапулезной сыпи с последующим образованием пузырей, имеющих тенденцию к слиянию. Наиболее частой была локализация высыпаний в области спины и боковых поверхностей туловища, на нижних конечностях, в паховой области, реже – на руках, волосистой части головы и лице. Во время высыпаний температура тела не повышалась. Соскобы содержимого пузырей у всех больных были стерильны. Во всех случаях на первой стадии заболевания возникли трудности дифференциальной диагностики. Только у одного ребенка в возрасте 10 дней дерматологом был предположен диагноз синдрома Блоха-Сульцбергера, в остальных случаях детям ставили следующие диагнозы: буллезный эпидермолиз, распространенная токсическая эритема новорожденных, герпетическая инфекция, стафило- и стрептодермия, тяжелая форма аллергии на белки грудного и коровьего молока. Отсутствие корректного диагноза в неонатальном периоде приводило к отсутствию адекватных терапевтических мер. Все дети получали массивную антибактериальную терапию, в некоторых случаях – противовирусные препараты (неовир, ацикловир), один ребенок – повторные курсы преднизолона. У 9 детей в периоде новорожденности было зарегистрировано повышение абсолютного и относительного количества эозинофилов периферической крови.
Течение первой стадии заболевания носило волнообразный характер с периодическим появлением новых высыпаний на непораженных участках кожи. У большинства больных эта фаза изменений кожи заканчивалась к 3–4 месяцам, а у отдельных детей – к 2м месяцам. У одного ребенка новые элементы сыпи продолжали появляться вплоть до 9 месяцев. Ещё у одной больной эритематозно-папулезной стадии не наблюдалось, по-видимому, она прошла внутриутробно. На коже волосистой части головы на местах высыпаний у детей оставались участки алопеции. Одновременно с появлением новых эритематозно-везикулезных элементов на месте старых высыпаний оставались папулы с гиперкератозом. Таким образом, переход ко второй стадии кожных изменений был постепенным. Вторая стадия наблюдалась нами только у одного 5-месячного ребенка, у половины детей её характеристику удалось восстановить ретроспективно по результатам анамнеза, в остальных случаях эта стадия прошла незамеченной либо отсутствовала. Начиная с возраста нескольких месяцев, у больных появлялись пигментные отложения, напоминающие «брызги грязи». Это третья стадия кожных изменений, которая наблюдалась нами у детей в возрасте от 10 месяцев до 8 лет. В этот период у одного из детей была проведена биопсия кожи нижней трети спины из участка пигментации, которая выявила скопления меланина в базальном и супрабазальном рядах эпителиоцитов, умеренную вакуолизацию базальных эпителиоцитов, умеренный отёк, свободные и внутриклеточные скопления пигмента. Примерно с пятилетнего возраста, наряду с остающимися пигментными отложениями, которые постепенно становились менее интенсивными, появлялись очаги депигментации, которые располагались по ходу линий роста кожи (линий Блашко), образуя характерный рисунок в виде «перьев». Четвертая стадия кожных изменений наблюдалась нами у детей в возрасте от 8 до 15 лет. Следует отметить постепенность перехода между стадиями. На рисунке 11 представлена последовательность изменений кожи у наблюдавшихся больных.
Анализ раннего развития детей показал, что у большинства из них была задержка становления двигательных навыков, кроме того наблюдалось отставание сроков появления слов и фразовой речи. Психическое развитие соответствовало возрасту только у одной девочки, у 11 детей наблюдались лёгкие когнитивные нарушения и у одной девочки – умеренная умственная отсталость. В неврологическом статусе отмечались разнообразные симптомы: нистагм, пирамидные нарушения, эпилептиформная активность на ЭЭГ, сочетание различных проявлений – судороги и спастический парапарез, сходящееся косоглазие и задержка моторного развития, судороги, пирамидная недостаточность и косоглазие, координаторные расстройства и дизартрия.
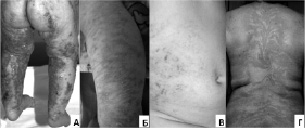
Рис.11. Стадии изменений кожи у наблюдавшихся больных с синдромом Блоха-Сульцбергера: А – эритематозно-везикулезные изменения,
Б – папулезные высыпания с гиперкератозом, В – пигментные отложения, Г – участки депигментации, соответствующие линиям Блашко
Таким образом, при обследовании детей с синдромом Блоха-Сульцбергера наблюдались изменения различных органов и систем. Физическое развитие, как правило, оценивалось как среднее гармоничное, обращало внимание уменьшение окружности головы у половины больных, которое у трети детей достигало степени микроцефалии. У значительной части детей выявлено отсутствие зубов, аномалии их формы и расположения (рис. 12, А). Патологические изменения других систем органов у больных с синдромом Блоха-Сульцбергера включали двустороннюю переднеполярную врожденную катаракту, аномалии почек, отсутствие ткани одной из молочных желёз, разнообразные аномалии скелета (рис. 12, Б). Обязательным фенотипическим признаком у больных с синдромом Блоха-Сульцбергера является характерное стадийное поражение кожи, остальные симптомы широко варьируют по частоте (когнитивные нарушения, аномалии скелета, глаз и др.) и степени выраженности.
Синдром Айкарди (OMIM 304050), как синдромы Блоха-Сульцбергера и RTT, наблюдается почти исключительно у девочек, поскольку для индивидуумов мужского пола характерна внутриутробная летальность. Известны отдельные случаи синдрома у мальчиков, имеющих лишнюю хромосому X в кариотипе (47,XXY) – синдром Клайнфельтера. До настоящего времени ген синдрома Айкарди не идентифицирован. Как и RTT, синдром Айкарди встречается спорадически, каждый его случай представляет собой результат мутации de novo. Единичные случаи синдрома Айкарди у сибсов предположительно являются результатом гонадного мозаицизма у родителей.
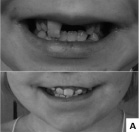
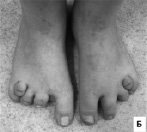
Рис. 12. Аномалии развития у детей с синдромом Блоха-Сульцбергера: А – аномалии зубов у девочек 12 и 8 лет, Б – аномалии пальцев стоп у ребенка 12 лет
Заболевание характеризуется триадой симптомов: частичным или полным отсутствием мозолистого тела, инфантильными спазмами и хориоретинальными лакунами. Другими изменениями мозга, определяемыми с помощью МРТ, могут быть микроцефалия, полимикрогирия, поренцефалические кисты, увеличение мозговых желудочков вследствие гидроцефалии. Манифестация синдрома Айкарди наступает до достижения ребенком возраста пяти месяцев (наиболее часто 3 мес.), когда впервые начинаются судороги, представленные, в основном, инфантильными спазмами. Наблюдаются специфические аномалии сетчатки глаз, известные как «лакуны», колобома дисков зрительных нервов, катаракта, нистагм. У всех детей отмечается грубая задержка психического и моторного развития. Встречаются аномалии скелета, такие как отсутствие или дополнительные ребра, расщелина нёба, аномалии позвонков и сколиоз. Могут наблюдаться изменения кожи в виде множественных невусов, гипопигментированных участков или гемангиом. Характерен ранний пубертатный период развития [Kroner et al., 2008]. Нами наблюдалась одна больная с синдромом Айкарди в возрасте 1 года. Девочка родилась в срок в ягодичном предлежании от первой беременности, протекавшей с угрозой прерывания, с массой тела 3150 г, длиной – 55 см. Неонатальный период протекал без патологии. Однако с трех месяцев появились эпилептические приступы в виде остановки взора с одномоментным вскидыванием рук вверх, которые постепенно сменились на «кивки», носившие серийный характер, появляющиеся перед сном или при просыпании. При обследовании в клинике в возрасте 1 года физическое развитие ребёнка было расценено как среднее гармоничное (длина тела – 78 см, масса тела – 10,5 кг, окружность головы – 46 см). В неврологическом статусе наблюдались гипотония мышц, инфантильные спазмы (4–5 серий приступов ежедневно) и выраженная задержка психомоторного развития (девочка удерживала голову, лёжа на животе, самостоятельно не сидела, не ползала и не ходила, произносила отдельные слоги). МРТ головного мозга выявила тотальную агенезию мозолистого тела с отсутствующей перикаллезной извилиной; кистозную трансформацию заднего рога левого бокового желудочка и деформацию передних рогов обоих боковых желудочков (рис. 13, А). При офтальмологическом обследовании обнаружены поражение зрительных путей, расширенная экскавация зрительного нерва, лакунарные хориоретинальные очаги, сложный гиперметропический астигматизм и амблиопия высокой степени (рис. 13, Б).
На основании триады клинических критериев – агенезия мозолистого тела, хориоретинальные лакуны и инфантильные спазмы – у ребёнка был установлен диагноз синдрома Айкарди. Несмотря на массивную комбинированную противосудорожную терапию, течение симптоматической эпилепсии у девочки было прогрессирующим, что привело её к смерти в эпилептическом статусе в возрасте 1 года 7 месяцев.
Фокальная дермальная гипоплазия Гольтца (OMIM 305 600) – мультисистемное заболевание, характеризующееся преимущественно поражением кожи, а также скелета и глаз. В основе заболевания лежат мутации гена PORCN. Большинство больных (90 %) женского пола являются гетерозиготами по мутации данного гена и 10 % больных – мальчики с мозаицизмом по мутации de novo. Регулярная мутация гена PORCN ведет к эмбриональной летальности у мужчин. Среди больных девочек у 95 % наблюдается новая мутация и в 5 % случаев она унаследована
от родителя [Lombardi et al., 2011]. Клинические проявления наблюдаются уже с рождения и представляют собой атрофические и гипопластические участки кожи с просвечивающими через них жировыми узелками, которые выглядят как желто-розовые субкутанные образования в коже. Наблюдаются нарушения пигментации кожи, гипоплазия ногтей, редкие волосы или их отсутствие. Встречаются аномалии кистей и стоп, в частности олиго/синдактилия. Аномалии глаз представлены микрофтальмией/анофтальмией, колобомой радужки и сетчатки, могут вовлекать назолакримальный канал. Особенности лица включают его асимметрию, зазубренные края крыльев носа, расщелину губы и неба и заостренный подбородок. Могут также наблюдаться аномалии зубов, дефекты брюшной стенки, диафрагмальные грыжи и патология почек. Психомоторное развитие нарушается не у всех детей: 15–20 % индивидуумов имеют нарушения интеллекта и около 20 % поведенческие проблемы в виде эмоциональной лабильности и аутистических черт, описаны единичные случаи эпилепсии [Goltz et al., 1962]. В биоптатах пораженных участков кожи у больных уменьшено количество коллагеновых и эластиновых волокон, повышено количество папиллярных дермальных кровеносных сосудов, уменьшена толщина дермы, фрагменты жировой ткани распределены по ней, наблюдаются папилломы [Ko et al., 2016].
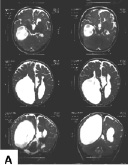
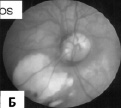
Рис. 13. Агенезия мозолистого тела, вентрикуломегалия, обнаруженные при магнитно-резонансной томографии головного мозга (А), и хориоретинальные лакуны, выявленные при офтальмоскопии (Б), у девочки с синдромом Айкарди
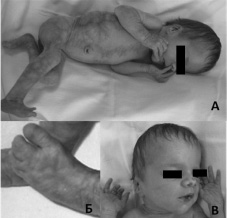
Рис. 14. Фенотип девочки 10-дневного возраста с фокальной дермальной гипоплазией Гольтца: А – очаги гипоплазии кожи, через которые выпячивается подкожная клетчатка, олигодактилия правой стопы,
Б – синдактилия левой стопы, В – телеангиэктазии на кончике носа,
заострённый подбородок, деформированные ушные раковины
Нами наблюдалась фокальная дермальная гипоплазия Гольтца у новорожденной девочки в возрасте 10 дней. Родители пробанда были здоровы, их возраст при рождении ребёнка был по 26 лет. Настоящая беременность была первой и протекала с угрозой прерывания. Роды произошли на 36 неделе, протекали стремительно. Девочка родилась с массой тела 2700 г, длиной – 48 см и окружностью головы – 33 см, все показатели соответствовали сроку гестации. При осмотре ребёнка на первый план выступали поражения кожи и её придатков: очаги гипоплазии кожи, выпячивания подкожной клетчатки через истонченную кожу, наиболее крупное – на правом боку, телеангиэктазии на кончике носа, волнистые гипопластичные ногтевые пластинки. Отмечались аномалии конечностей в виде олигодактилии правой стопы и полной синдактилии II–III пальцев левой стопы, укорочения II пальца левой кисти. Обращали на себя внимание лицевые микроаномалии: башенной формы череп, асимметрия лица (левая половина меньше правой), заострённый подбородок, низкорасположенные деформированные ушные раковины (рис. 14). При офтальмологическом обследовании выявлены распространённый отёк и аваскулярные зоны на сетчатке обоих глаз. Нейросонография, проведённая на 6-й день жизни, обнаружила расширение переднего рога и тела правого бокового желудочка. Ультразвуковое исследование сердца, органов брюшной полости и почек патологии не выявило. Диагноз фокальной дермальной гипоплазии был поставлен ребенку на основании клинических критериев: типичных очагов гипоплазии кожи, аномалий скелета в виде пороков развития стоп и характерных лицевых микроаномалий.
Таким образом, заболеваниям, проявляющимся у гетерозигот, обычно свойственна стадийность (синдромы Блоха-Сульцбергера, RTT), прогрессирующий характер (синдромы Айкарди и RTT). Смена стадий может отражать процесс взаимодействия клеток с активными мутантной и нормальной хромосомами Х у гетерозигот. В частности, стадии поражения кожи при синдроме Блоха-Сульцбергера, в особенности острая первая стадия, являются проявлением гибели клеток с активной мутантной хромосомой Х.
При фокальной дермальной гипоплазии Гольтца гибель клеток кожи с активной хромосомой Х, несущей мутацию, по-видимому, проходит внутриутробно, поэтому к моменту рождения пробанда наблюдаются сформированные дефекты кожи. Очаги поражения сетчатки и пороки развития мозга при синдроме Айкарди, вероятно, также возникают в результате селекции клеток. Данному синдрому свойственно быстрое прогрессирование заболевания, заканчивающееся гибелью ребенка в первые годы жизни. Можно предположить, что процесс селекции клеток лежит в основе острой стадии регресса при RTT. У мальчиков-гемизигот с XLMR обычно не наблюдается стадийности течения заболевания. При метаболических болезнях (мукополисахаридоз II типа) болезнь прогрессирует постепенно (без острой стадии) вследствие нарастания метаболических нарушений.
Характерной особенностью XLMR является выраженный клинический полиморфизм, который в значительной степени затрудняет диагностику данной патологии.